

The landscape of coding RNA editing events in pediatric cancer
- PMID: 34789196
- PMCID: PMC8597231
- DOI: 10.1186/s12885-021-08956-5
The landscape of coding RNA editing events in pediatric cancer
Abstract
Background: RNA editing leads to post-transcriptional variation in protein sequences and has important biological implications. We sought to elucidate the landscape of RNA editing events across pediatric cancers.
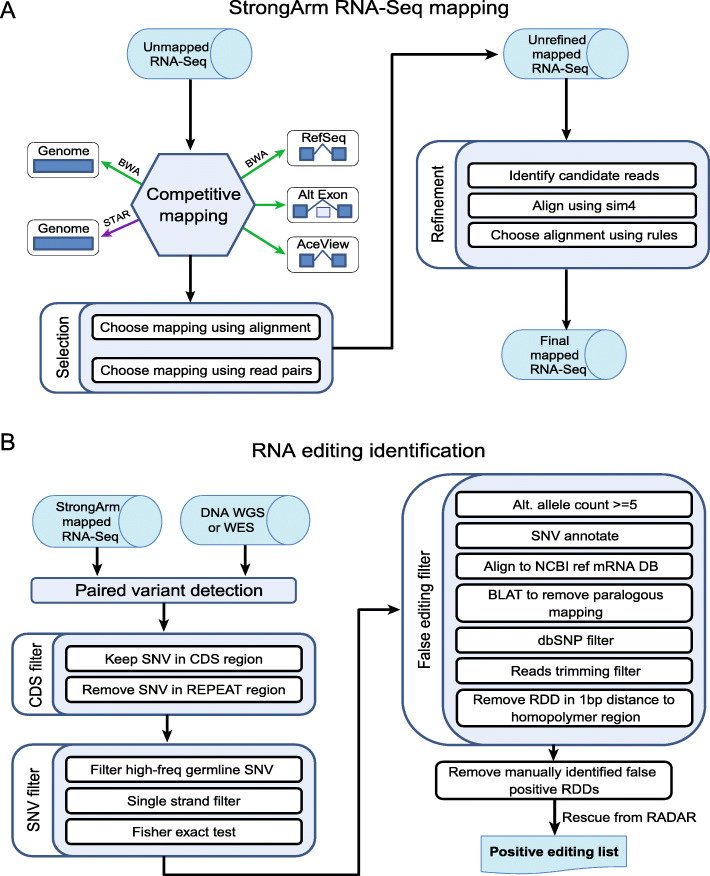
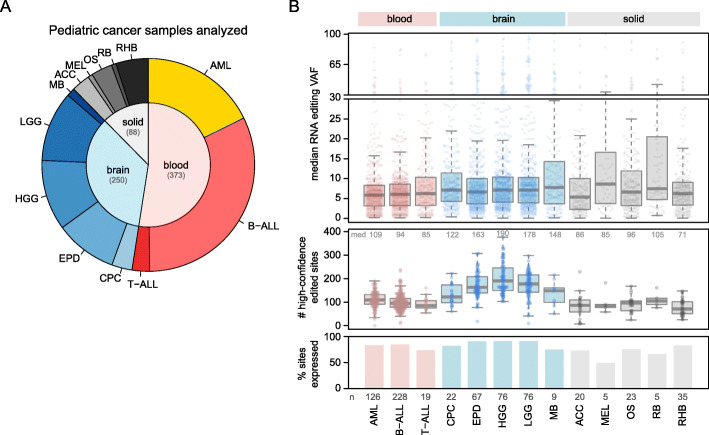
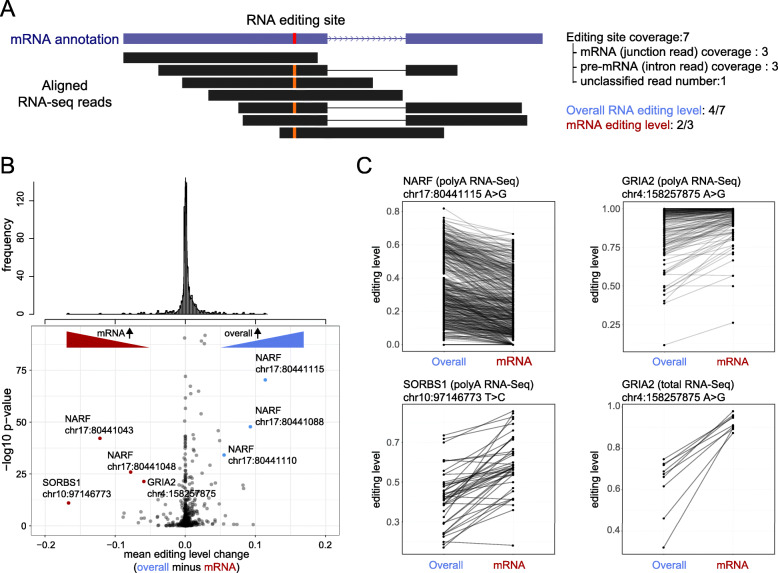
Methods: Using RNA-Seq data mapped by a pipeline designed to minimize mapping ambiguity, we investigated RNA editing in 711 pediatric cancers from the St. Jude/Washington University Pediatric Cancer Genome Project focusing on coding variants which can potentially increase protein sequence diversity. We combined de novo detection using paired tumor DNA-RNA data with analysis of known RNA editing sites.
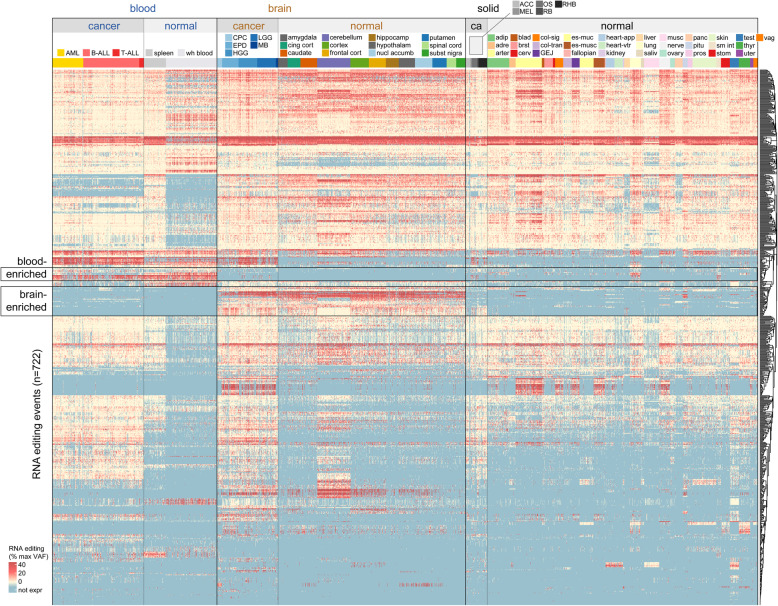
Results: We identified 722 unique RNA editing sites in coding regions across pediatric cancers, 70% of which were nonsynonymous recoding variants. Nearly all editing sites represented the canonical A-to-I (n = 706) or C-to-U sites (n = 14). RNA editing was enriched in brain tumors compared to other cancers, including editing of glutamate receptors and ion channels involved in neurotransmitter signaling. RNA editing profiles of each pediatric cancer subtype resembled those of the corresponding normal tissue profiled by the Genotype-Tissue Expression (GTEx) project.
Conclusions: In this first comprehensive analysis of RNA editing events in pediatric cancer, we found that the RNA editing profile of each cancer subtype is similar to its normal tissue of origin. Tumor-specific RNA editing events were not identified indicating that successful immunotherapeutic targeting of RNA-edited peptides in pediatric cancer should rely on increased antigen presentation on tumor cells compared to normal but not on tumor-specific RNA editing per se.
Keywords: Genomics; Immunotherapy; Pediatric cancer; RNA editing.
© 2021. The Author(s).
Conflict of interest statement
CGM has received consulting and speaking fees from Illumina and Amgen, and research support from Loxo Oncology, Pfizer and Abbvie, and holds stock in Amgen.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
