

Independent infections of porcine deltacoronavirus among Haitian children
- PMID: 34789872
- PMCID: PMC8636265
- DOI: 10.1038/s41586-021-04111-z
Independent infections of porcine deltacoronavirus among Haitian children
Abstract
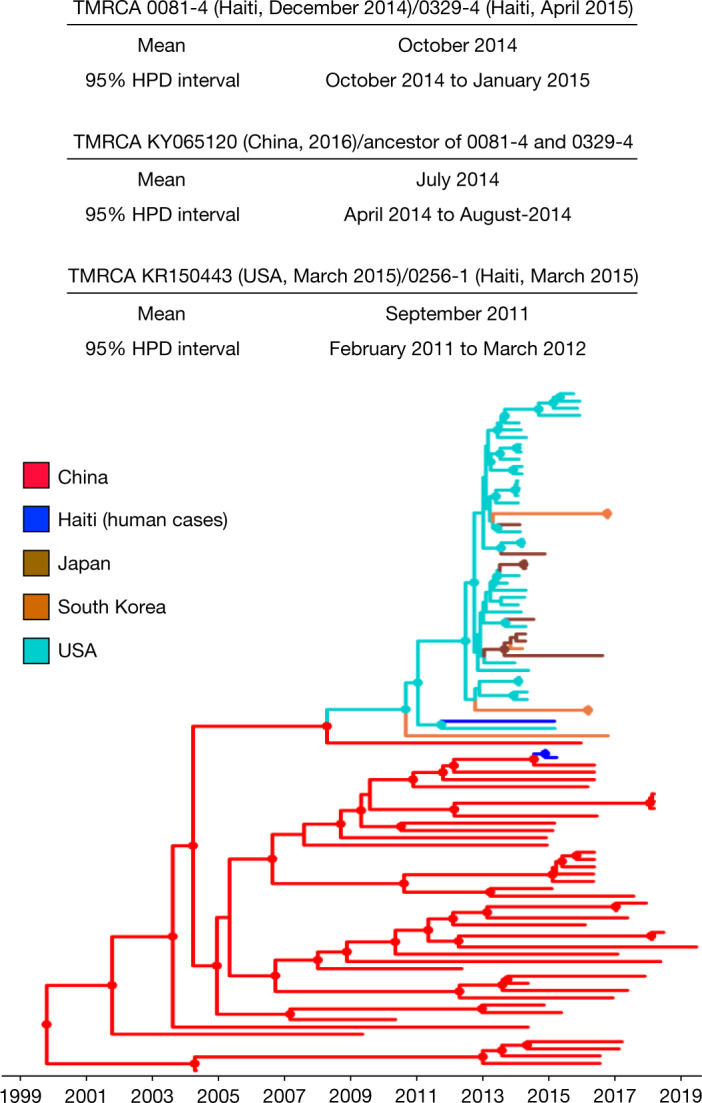
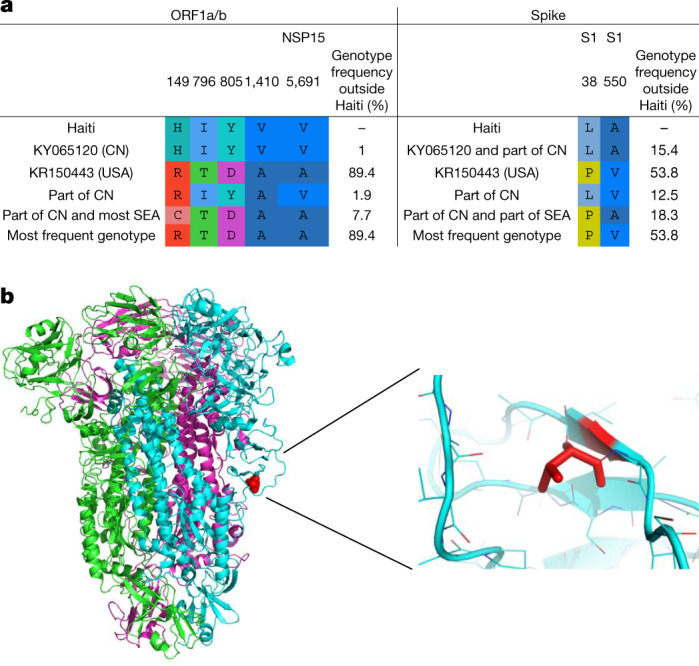
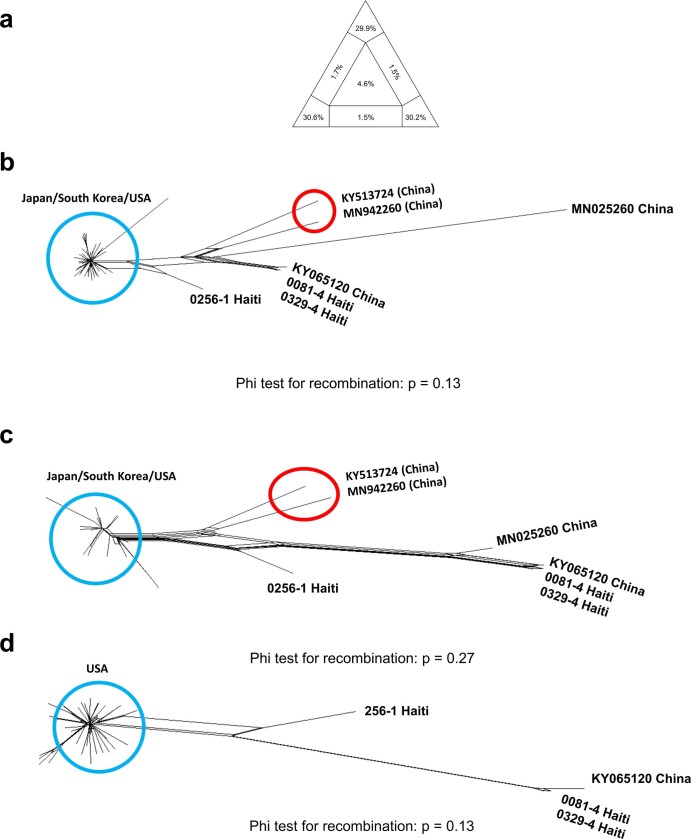
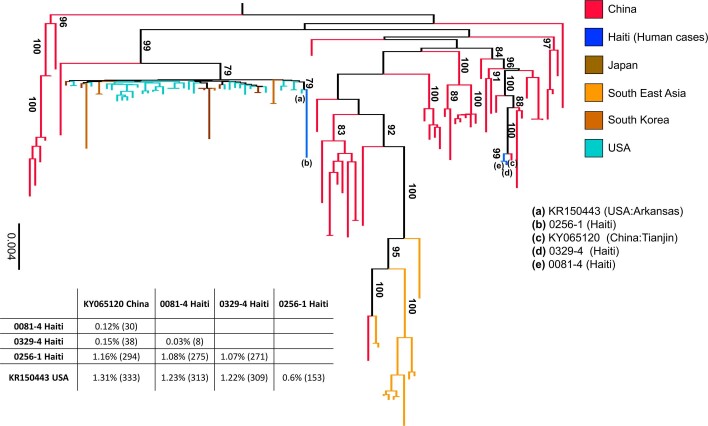
Coronaviruses have caused three major epidemics since 2003, including the ongoing SARS-CoV-2 pandemic. In each case, the emergence of coronavirus in our species has been associated with zoonotic transmissions from animal reservoirs1,2, underscoring how prone such pathogens are to spill over and adapt to new species. Among the four recognized genera of the family Coronaviridae, human infections reported so far have been limited to alphacoronaviruses and betacoronaviruses3-5. Here we identify porcine deltacoronavirus strains in plasma samples of three Haitian children with acute undifferentiated febrile illness. Genomic and evolutionary analyses reveal that human infections were the result of at least two independent zoonoses of distinct viral lineages that acquired the same mutational signature in the genes encoding Nsp15 and the spike glycoprotein. In particular, structural analysis predicts that one of the changes in the spike S1 subunit, which contains the receptor-binding domain, may affect the flexibility of the protein and its binding to the host cell receptor. Our findings highlight the potential for evolutionary change and adaptation leading to human infections by coronaviruses outside of the previously recognized human-associated coronavirus groups, particularly in settings where there may be close human-animal contact.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


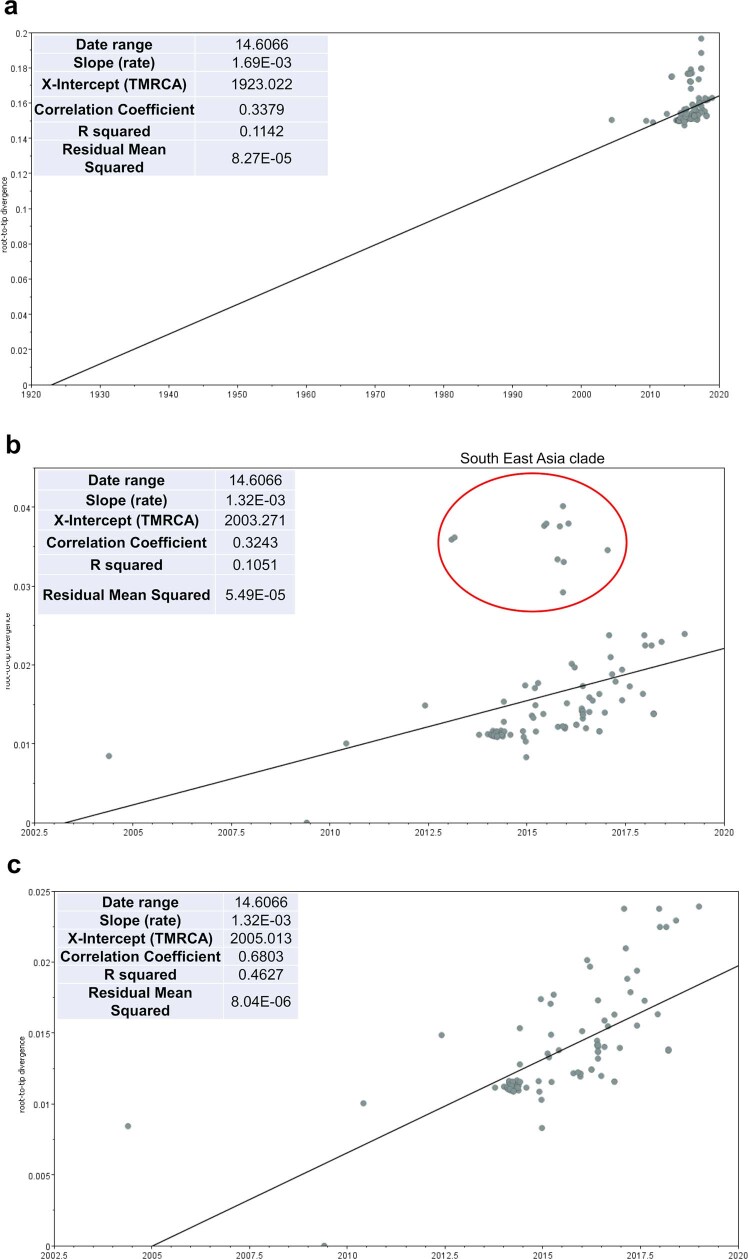
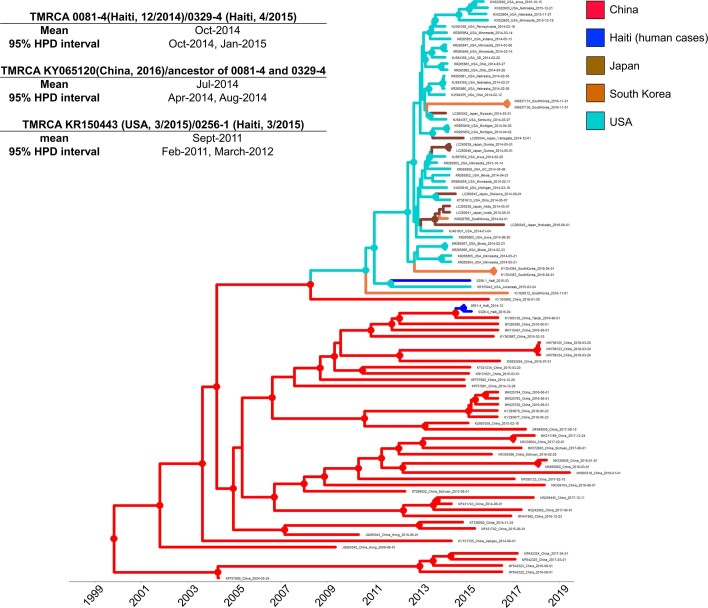
Update of
-
Emergence of porcine delta-coronavirus pathogenic infections among children in Haiti through independent zoonoses and convergent evolution.medRxiv [Preprint]. 2021 Mar 25:2021.03.19.21253391. doi: 10.1101/2021.03.19.21253391. medRxiv. 2021. Update in: Nature. 2021 Dec;600(7887):133-137. doi: 10.1038/s41586-021-04111-z. PMID: 33791709 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
