

Cell-type specialization is encoded by specific chromatin topologies
- PMID: 34789882
- PMCID: PMC8612935
- DOI: 10.1038/s41586-021-04081-2
Cell-type specialization is encoded by specific chromatin topologies
Abstract
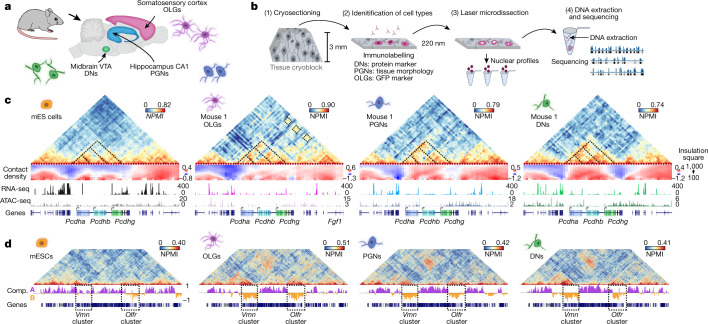
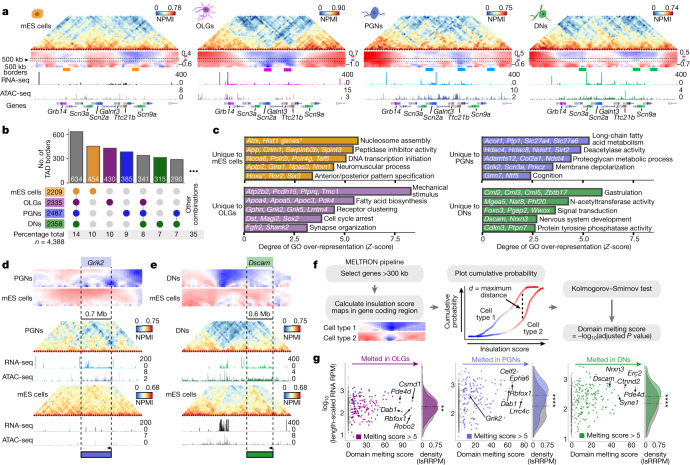
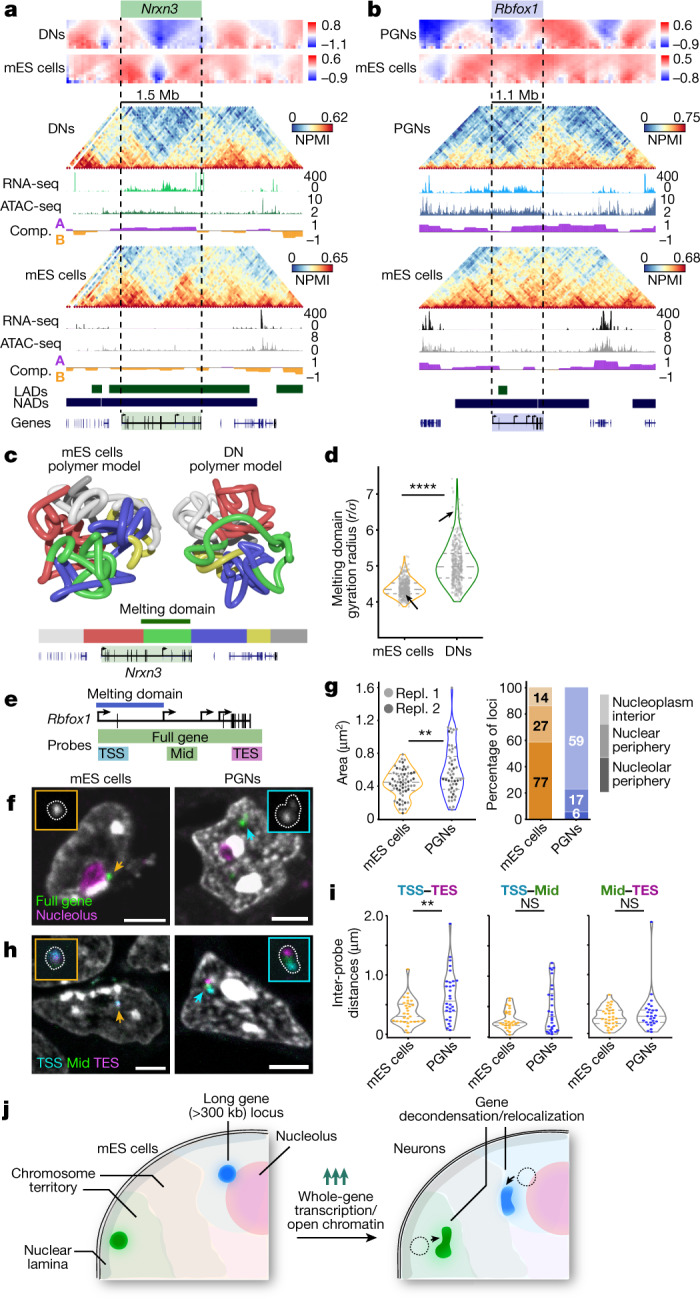
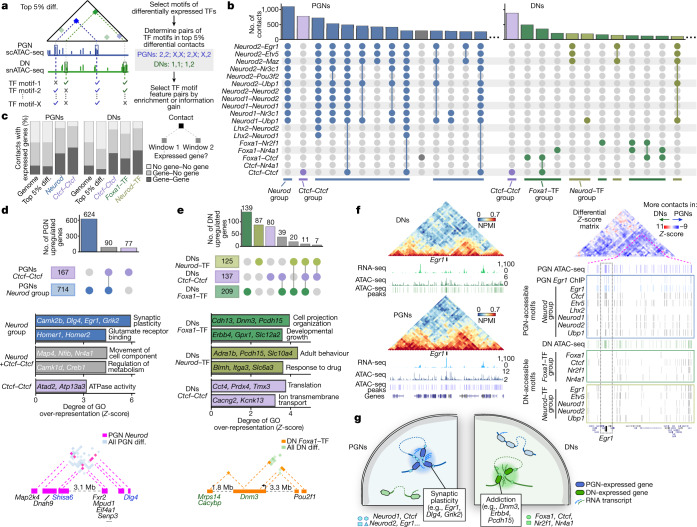
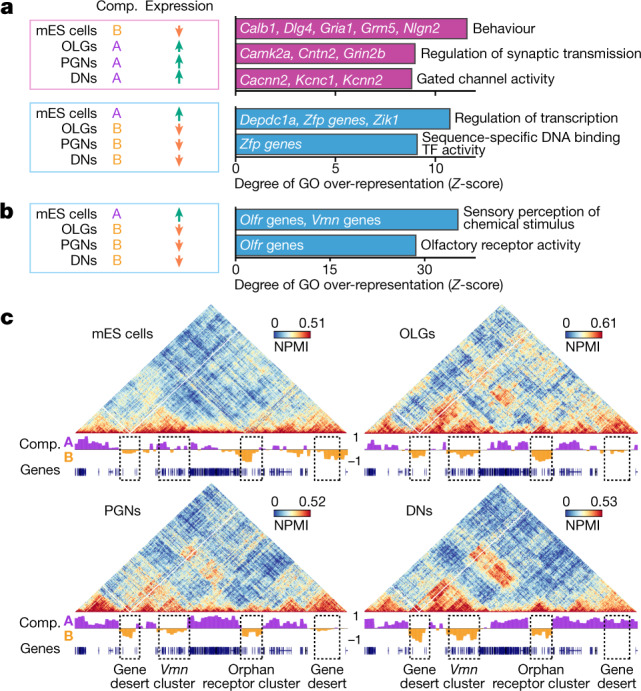
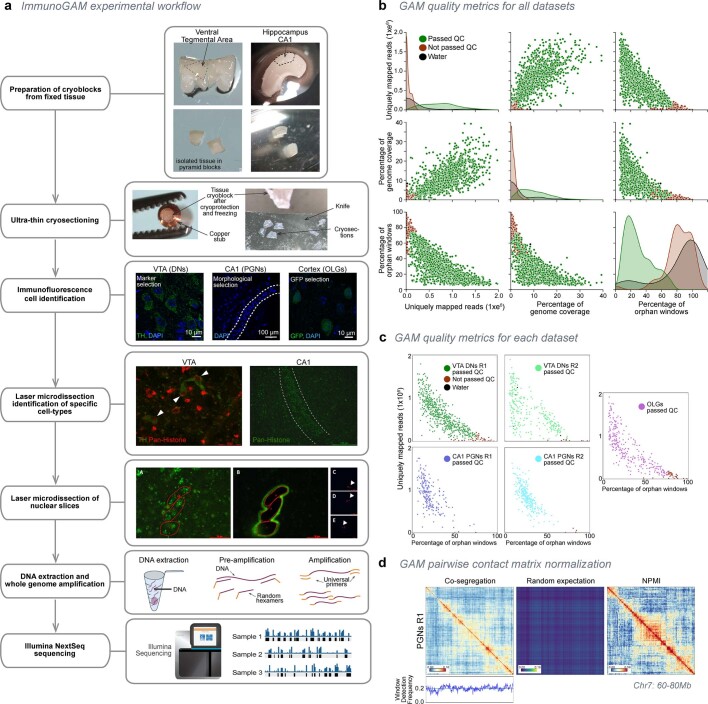
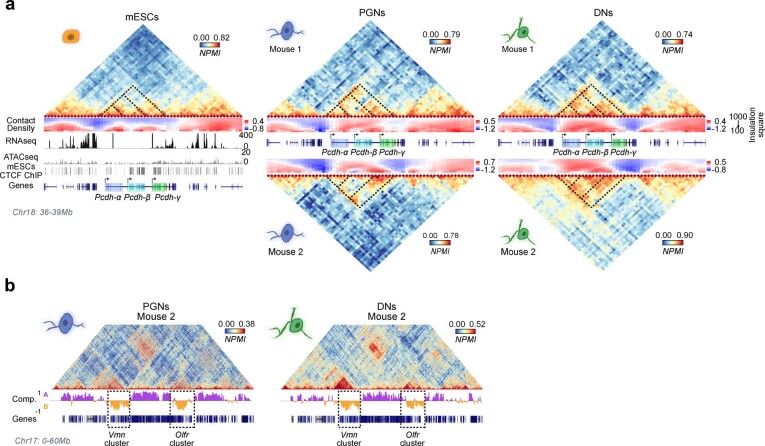
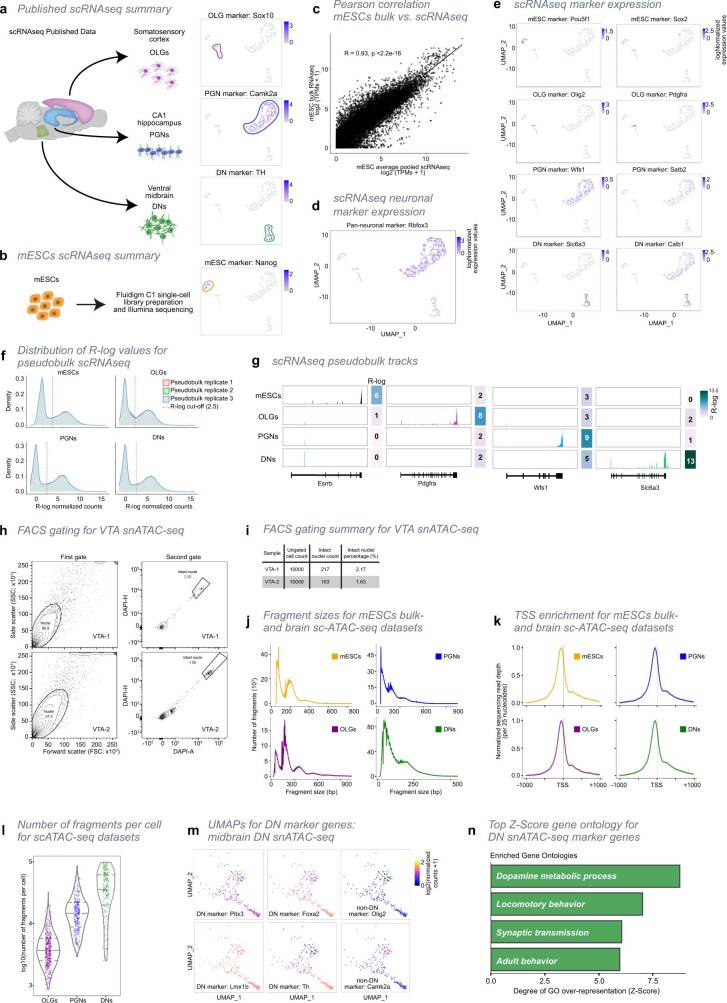
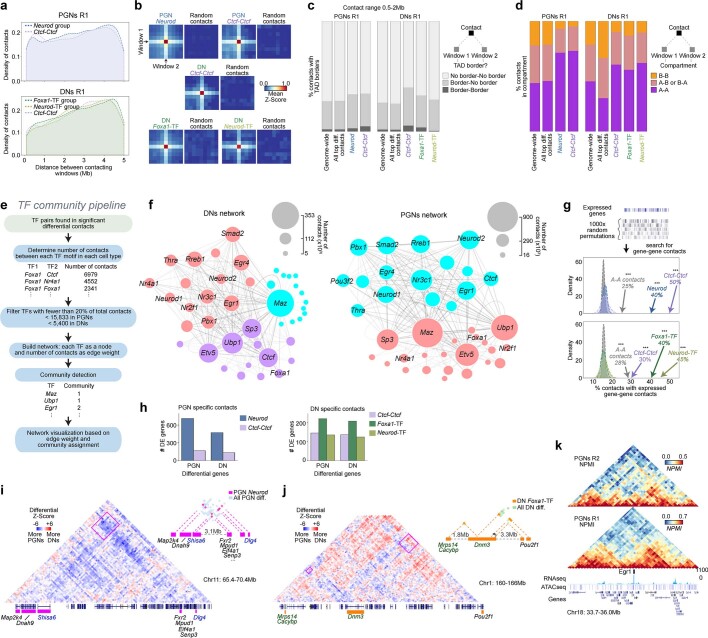
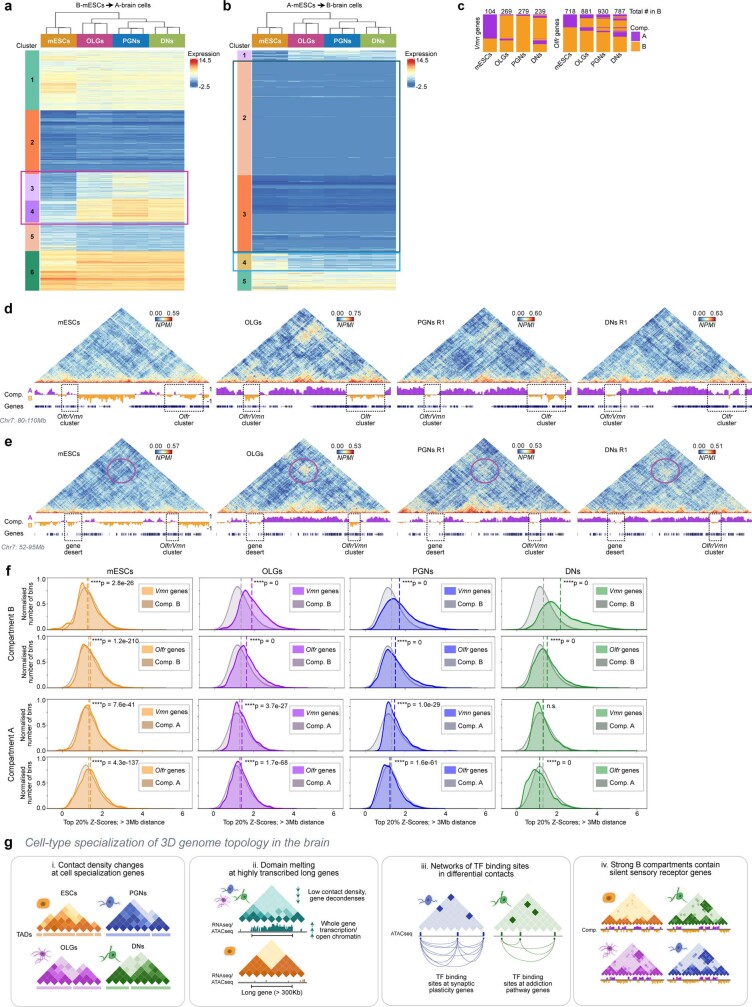
The three-dimensional (3D) structure of chromatin is intrinsically associated with gene regulation and cell function1-3. Methods based on chromatin conformation capture have mapped chromatin structures in neuronal systems such as in vitro differentiated neurons, neurons isolated through fluorescence-activated cell sorting from cortical tissues pooled from different animals and from dissociated whole hippocampi4-6. However, changes in chromatin organization captured by imaging, such as the relocation of Bdnf away from the nuclear periphery after activation7, are invisible with such approaches8. Here we developed immunoGAM, an extension of genome architecture mapping (GAM)2,9, to map 3D chromatin topology genome-wide in specific brain cell types, without tissue disruption, from single animals. GAM is a ligation-free technology that maps genome topology by sequencing the DNA content from thin (about 220 nm) nuclear cryosections. Chromatin interactions are identified from the increased probability of co-segregation of contacting loci across a collection of nuclear slices. ImmunoGAM expands the scope of GAM to enable the selection of specific cell types using low cell numbers (approximately 1,000 cells) within a complex tissue and avoids tissue dissociation2,10. We report cell-type specialized 3D chromatin structures at multiple genomic scales that relate to patterns of gene expression. We discover extensive 'melting' of long genes when they are highly expressed and/or have high chromatin accessibility. The contacts most specific of neuron subtypes contain genes associated with specialized processes, such as addiction and synaptic plasticity, which harbour putative binding sites for neuronal transcription factors within accessible chromatin regions. Moreover, sensory receptor genes are preferentially found in heterochromatic compartments in brain cells, which establish strong contacts across tens of megabases. Our results demonstrate that highly specific chromatin conformations in brain cells are tightly related to gene regulation mechanisms and specialized functions.
© 2021. The Author(s).
Conflict of interest statement
In the past 3 years, S.A.T. has acted as a consultant for Genentech and Roche, and is a remunerated member of Scientific Advisory Boards of Biogen, GlaxoSmithKline and Foresite Labs. A.P. and M.N. hold a patent on GAM.
Figures



Comment in
-
Cell type-specific chromatin topology and gene regulation.Trends Genet. 2022 May;38(5):413-415. doi: 10.1016/j.tig.2022.02.008. Epub 2022 Feb 24. Trends Genet. 2022. PMID: 35221113 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
