

Reinforcement of CHH methylation through RNA-directed DNA methylation ensures sexual reproduction in rice
- PMID: 34791444
- PMCID: PMC8825330
- DOI: 10.1093/plphys/kiab531
Reinforcement of CHH methylation through RNA-directed DNA methylation ensures sexual reproduction in rice
Abstract
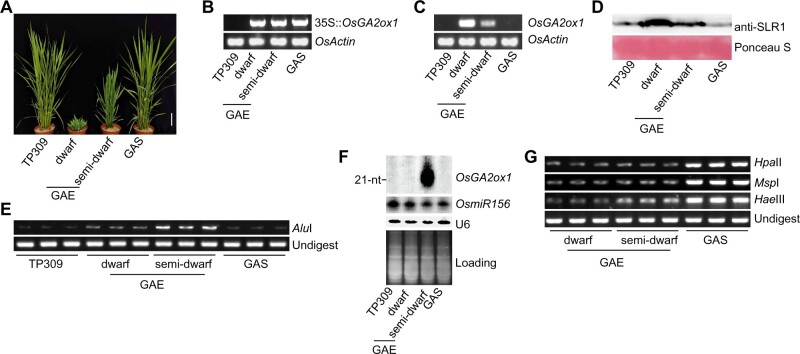
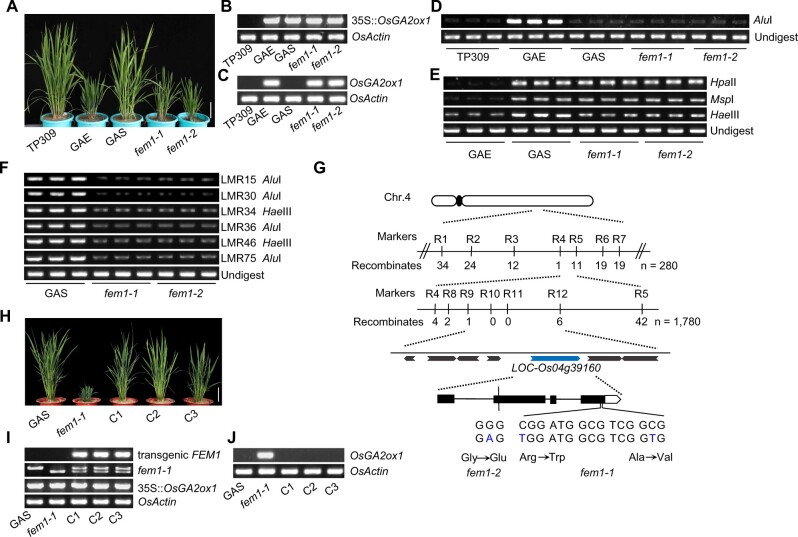
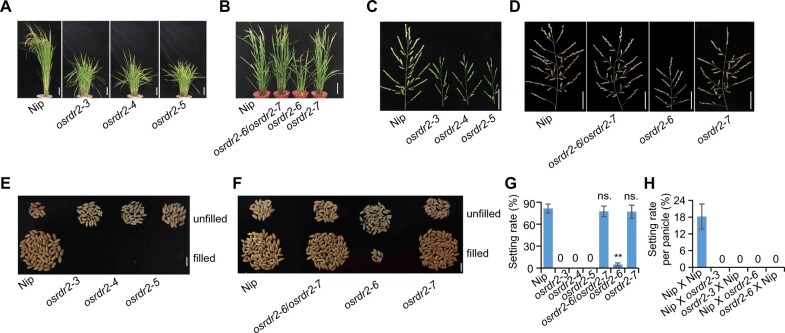
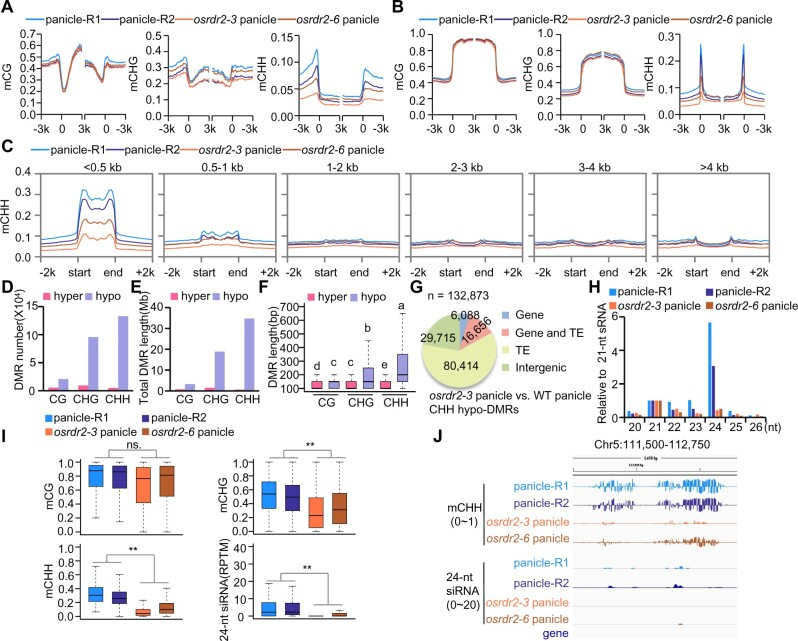
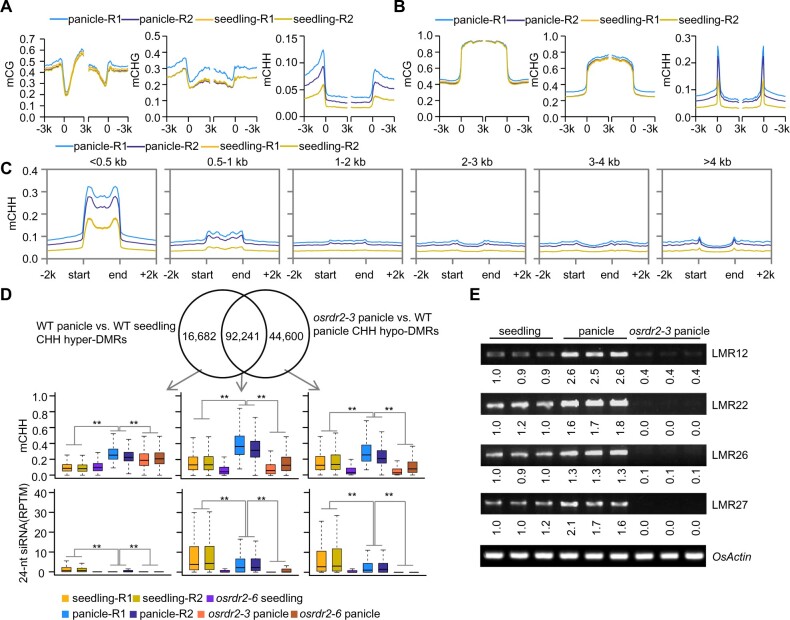
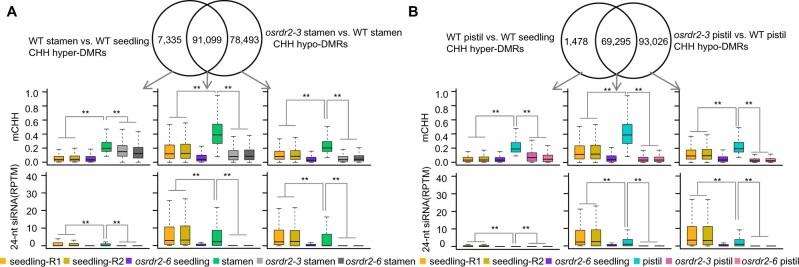
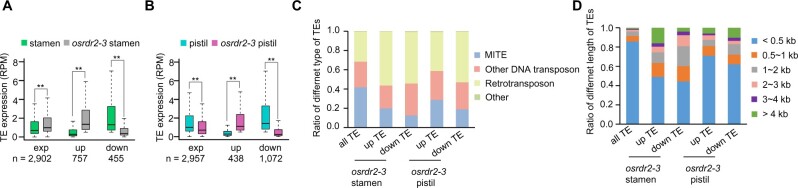
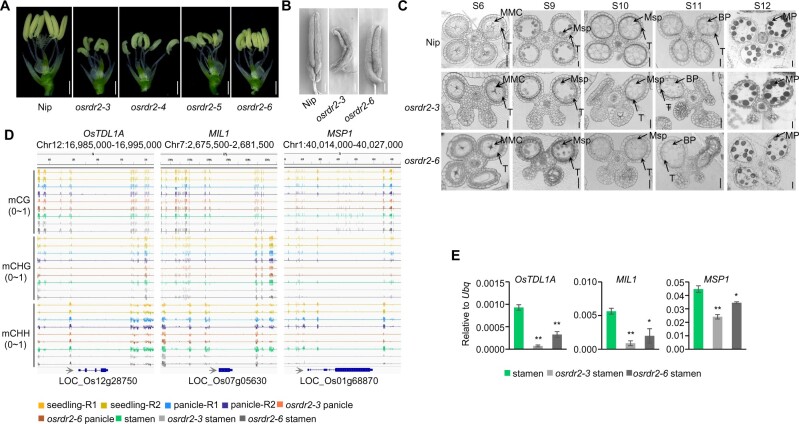
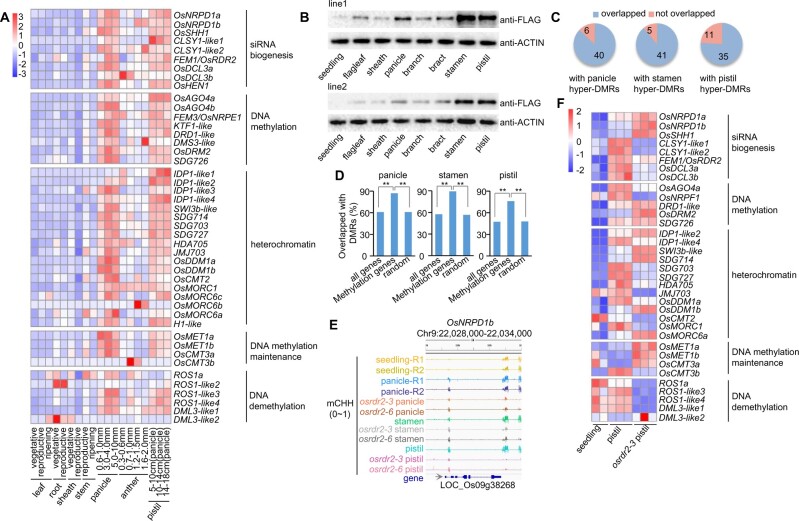
DNA methylation is an important epigenetic mark that regulates the expression of genes and transposons. RNA-directed DNA methylation (RdDM) is the main molecular pathway responsible for de novo DNA methylation in plants. Although the mechanism of RdDM has been well studied in Arabidopsis (Arabidopsis thaliana), most mutations in RdDM genes cause no remarkable developmental defects in Arabidopsis. Here, we isolated and cloned Five Elements Mountain 1 (FEM1), which encodes RNA-dependent RNA polymerase 2 (OsRDR2) in rice (Oryza sativa). Mutation in OsRDR2 abolished the accumulation of 24-nt small interfering RNAs, and consequently substantially decreased genome-wide CHH (H = A, C, or T) methylation. Moreover, male and female reproductive development was disturbed, which led to sterility in osrdr2 mutants. We discovered that OsRDR2-dependent DNA methylation may regulate the expression of multiple key genes involved in stamen development, meiosis, and pollen viability. In wild-type (WT) plants but not in osrdr2 mutants, genome-wide CHH methylation levels were greater in panicles, stamens, and pistils than in seedlings. The global increase of CHH methylation in reproductive organs of the WT was mainly explained by the enhancement of RdDM activity, which includes OsRDR2 activity. Our results, which revealed a global increase in CHH methylation through enhancement of RdDM activity in reproductive organs, suggest a crucial role for OsRDR2 in the sexual reproduction of rice.
© American Society of Plant Biologists 2021. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Alleman M, Sidorenko L, McGinnis K, Seshadri V, Dorweiler JE, White J, Sikkink K, Chandler VL (2006) An RNA-dependent RNA polymerase is required for paramutation in maize. Nature 442: 295–298 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
