

Ataluren delays loss of ambulation and respiratory decline in nonsense mutation Duchenne muscular dystrophy patients
- PMID: 34791888
- PMCID: PMC8787621
- DOI: 10.2217/cer-2021-0196
Ataluren delays loss of ambulation and respiratory decline in nonsense mutation Duchenne muscular dystrophy patients
Abstract
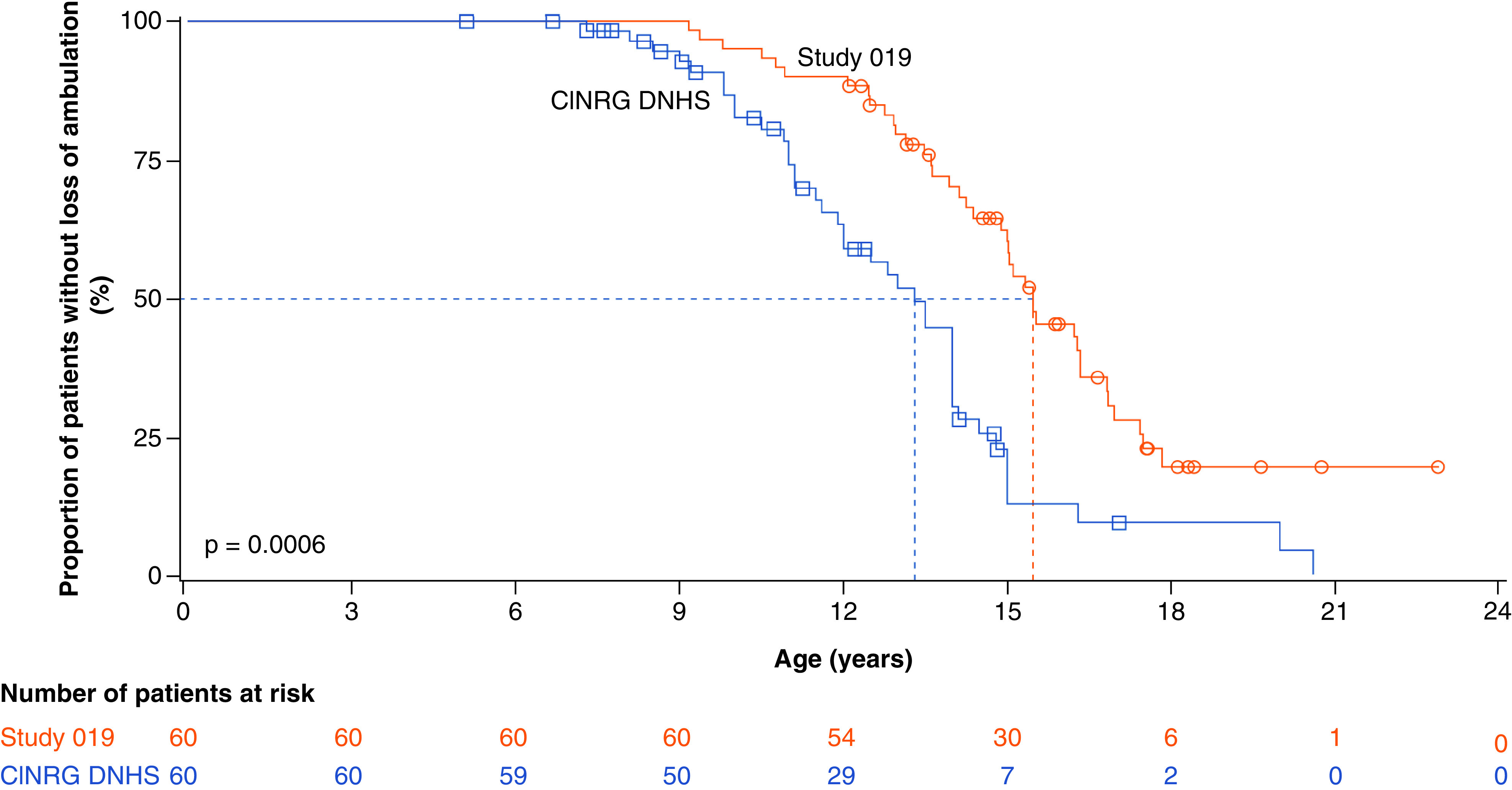
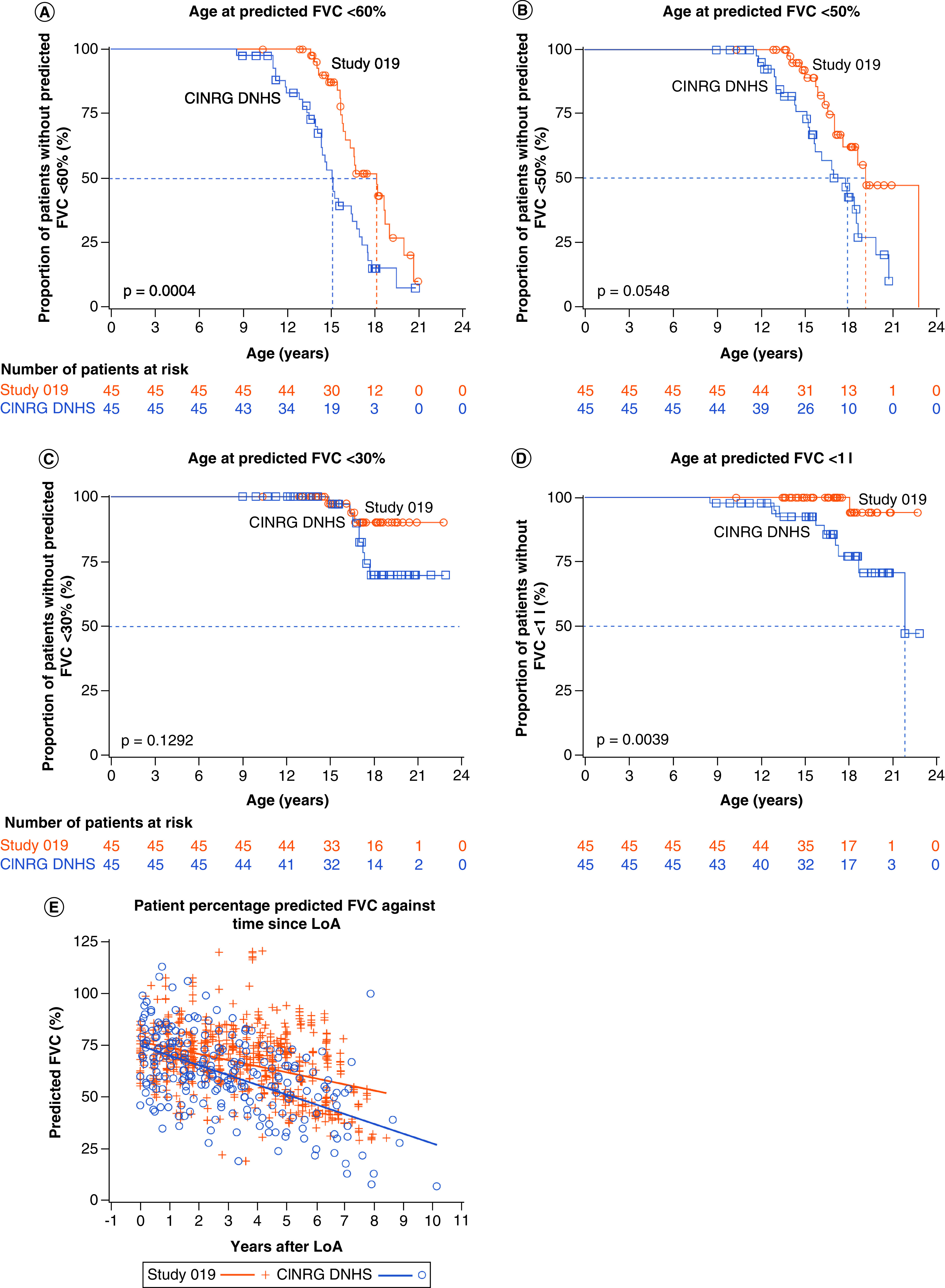
Aim: We investigated the effect of ataluren plus standard of care (SoC) on age at loss of ambulation (LoA) and respiratory decline in patients with nonsense mutation Duchenne muscular dystrophy (nmDMD) versus patients with DMD on SoC alone. Patients & methods: Study 019 was a long-term Phase III study of ataluren safety in nmDMD patients with a history of ataluren exposure. Propensity score matching identified Study 019 and CINRG DNHS patients similar in disease progression predictors. Results & conclusion: Ataluren plus SoC was associated with a 2.2-year delay in age at LoA (p = 0.0006), and a 3.0-year delay in decline of predicted forced vital capacity to <60% in nonambulatory patients (p = 0.0004), versus SoC. Ataluren plus SoC delays disease progression and benefits ambulatory and nonambulatory patients with nmDMD. ClinicalTrials.gov registration: NCT01557400.
Keywords: Study 019; ataluren; dystrophin; efficacy; loss of ambulation; nonsense mutation Duchenne muscular dystrophy; respiratory function.
Conflict of interest statement
This work was supported by PTC Therapeutics. Study 019 was sponsored by PTC Therapeutics. The CINRG DNHS was funded by grants from the US Department of Education/NIDRR (#H133B031118, #H133B090001), the US Department of Defense (#W81XWH-09-1-0592), the National Institutes of Health (#UL1RR031988, #U54HD053177, #UL1RR024992, #U54RR026139, #2U54HD053177, #G12RR003051, #1R01AR061875, #RO1AR062380) and Parent Project Muscular Dystrophy. CM McDonald has acted as a consultant on clinical trials of DMD for Astellas, BioMarin, Capricor Therapeutics, Catabasis Pharmaceuticals, Edgewise Therapeutics, Eli Lilly and company, Epirium Bio (formerly Cardero Therapeutics), FibroGen, Gilead, Hoffmann-La Roche, Italfarmaco, Pfizer, PTC Therapeutics, Santhera Pharmaceuticals and Sarepta Therapeutics, and has received research support for clinical trials from BioMarin, Capricor Therapeutics, Eli Lilly and company, Italfarmaco, Pfizer, PTC Therapeutics, Hoffmann-La Roche, Santhera Pharmaceuticals and Sarepta Therapeutics. F Muntoni has received consultancy fees from AveXis, Biogen, Capricor Therapeutics, Catabasis Pharmaceuticals, Dyne Therapeutics, Novartis, Pfizer, PTC Therapeutics, Roche, Santhera Pharmaceuticals, Sarepta Therapeutics and Wave Therapeutics, and is supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children National Health Service Foundation Trust and University College London. V Penematsa, J Jiang, A Kristensen, E Goodwin, C Werner, James Li, R Able and P Trifillis are employees of PTC Therapeutics. F Bibbiani is a former employee of PTC Therapeutics. H Gordish-Dressman has served as a consultant for AGADA Biosciences, Audentes Therapeutics, ReveraGen BioPharma and Solid GT, and is a cofounder and part owner of TRiNDS. L Morgenroth reports no disclosures relevant to this manuscript. M Tulinius has received lecture fees from Biogen and PTC Therapeutics, has acted as a consultant on DMD clinical trials for BioMarin, Catabasis Pharmaceuticals, PTC Therapeutics, ReveraGen BioPharma and Sarepta Therapeutics, and as an advisory board member for AveXis, Biogen, PTC Therapeutics and Sarepta Therapeutics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing and editorial support were provided by E Colbeck, PhD, an employee of PharmaGenesis London, London, UK, and were funded by PTC Therapeutics.
Figures

References
-
- Bushby K, Finkel R, Birnkrant DJ et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9(1), 77–93 (2010a). - PubMed
-
- Ellis JA, Vroom E, Muntoni F. 195th ENMC International Workshop: newborn screening for Duchenne muscular dystrophy 14–16th December, 2012, Naarden, The Netherlands. Neuromusc. Disord. 23(8), 682–689 (2013). - PubMed