

From genotype to phenotype in Arabidopsis thaliana: in-silico genome interpretation predicts 288 phenotypes from sequencing data
- PMID: 34792168
- PMCID: PMC8860592
- DOI: 10.1093/nar/gkab1099
From genotype to phenotype in Arabidopsis thaliana: in-silico genome interpretation predicts 288 phenotypes from sequencing data
Abstract
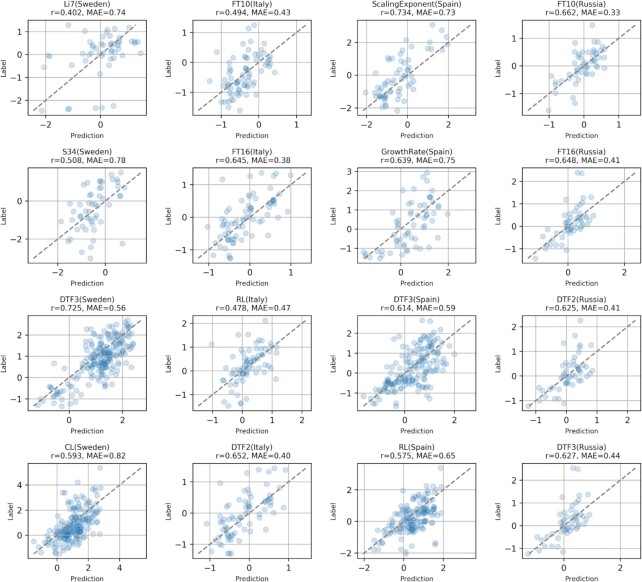
In many cases, the unprecedented availability of data provided by high-throughput sequencing has shifted the bottleneck from a data availability issue to a data interpretation issue, thus delaying the promised breakthroughs in genetics and precision medicine, for what concerns Human genetics, and phenotype prediction to improve plant adaptation to climate change and resistance to bioagressors, for what concerns plant sciences. In this paper, we propose a novel Genome Interpretation paradigm, which aims at directly modeling the genotype-to-phenotype relationship, and we focus on A. thaliana since it is the best studied model organism in plant genetics. Our model, called Galiana, is the first end-to-end Neural Network (NN) approach following the genomes in/phenotypes out paradigm and it is trained to predict 288 real-valued Arabidopsis thaliana phenotypes from Whole Genome sequencing data. We show that 75 of these phenotypes are predicted with a Pearson correlation ≥0.4, and are mostly related to flowering traits. We show that our end-to-end NN approach achieves better performances and larger phenotype coverage than models predicting single phenotypes from the GWAS-derived known associated genes. Galiana is also fully interpretable, thanks to the Saliency Maps gradient-based approaches. We followed this interpretation approach to identify 36 novel genes that are likely to be associated with flowering traits, finding evidence for 6 of them in the existing literature.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures



References
-
- Daneshjou R., Wang Y., Bromberg Y., Bovo S., Martelli P.L., Babbi G., Lena P.D., Casadio R., Edwards M., Gifford D.et al.. Working toward precision medicine: Predicting phenotypes from exomes in the Critical Assessment of Genome Interpretation (CAGI) challenges. Hum. Mutat. 2017; 38:1182–1192. - PMC - PubMed
-
- Moreau Y., Tranchevent L.-C.. Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat. Rev. Genet. 2012; 13:523–536. - PubMed
