

Single-Cell, Single-Nucleus, and Spatial RNA Sequencing of the Human Liver Identifies Cholangiocyte and Mesenchymal Heterogeneity
- PMID: 34792289
- PMCID: PMC8948611
- DOI: 10.1002/hep4.1854
Single-Cell, Single-Nucleus, and Spatial RNA Sequencing of the Human Liver Identifies Cholangiocyte and Mesenchymal Heterogeneity
Abstract
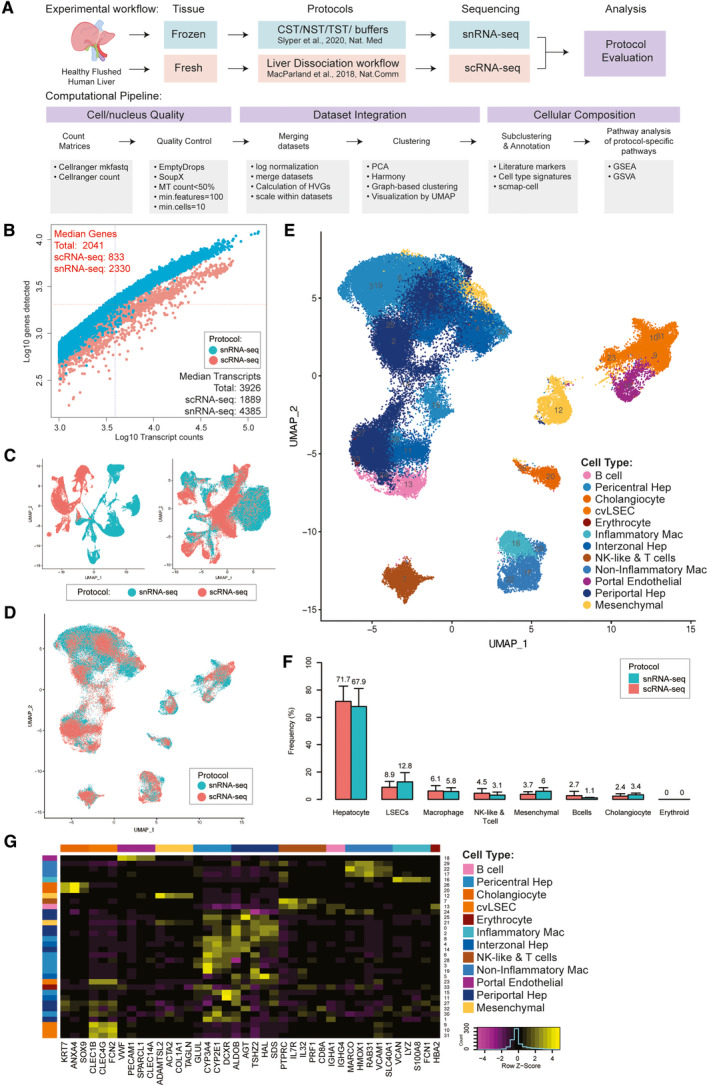
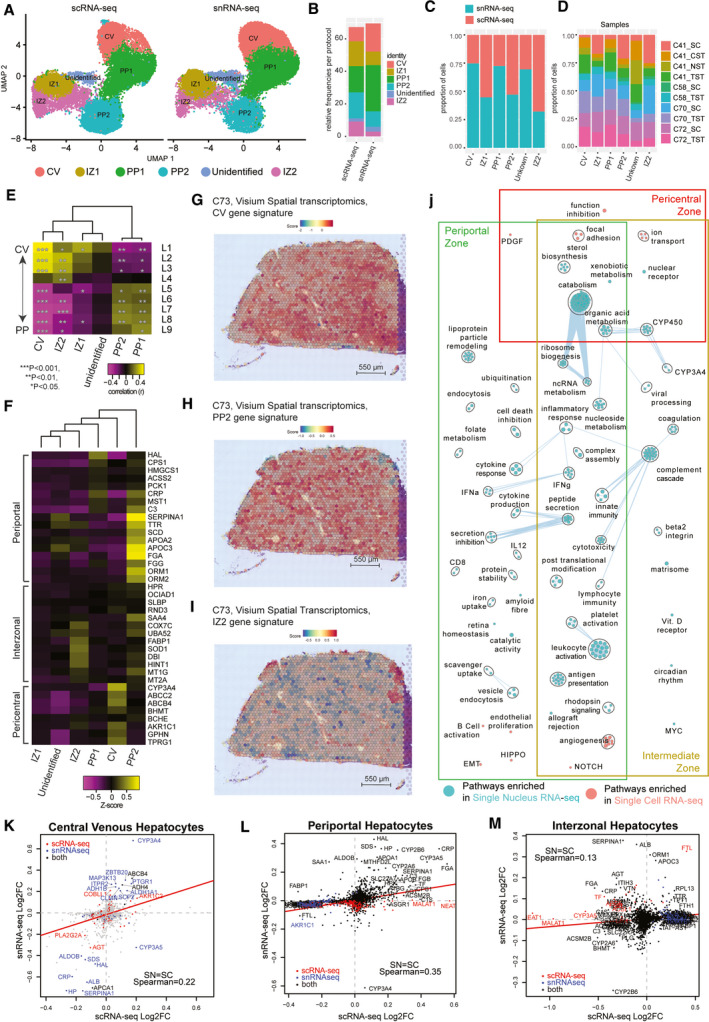
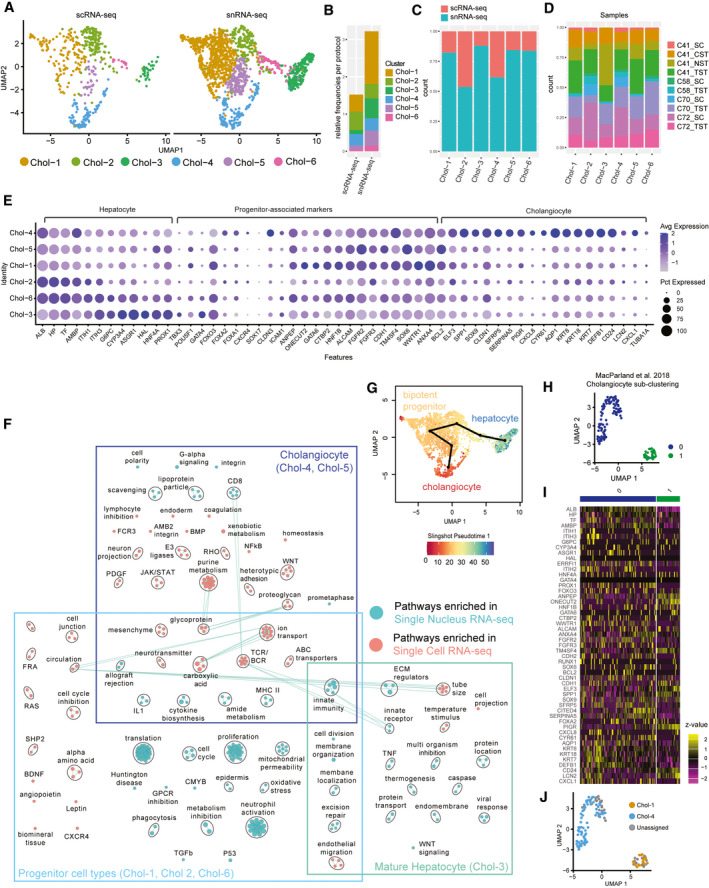
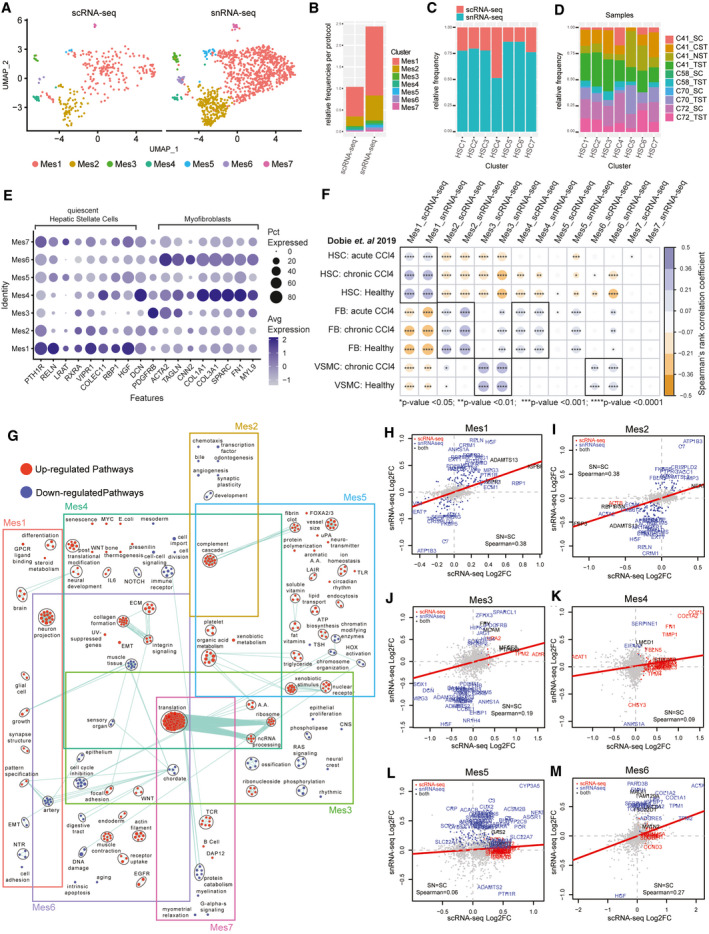
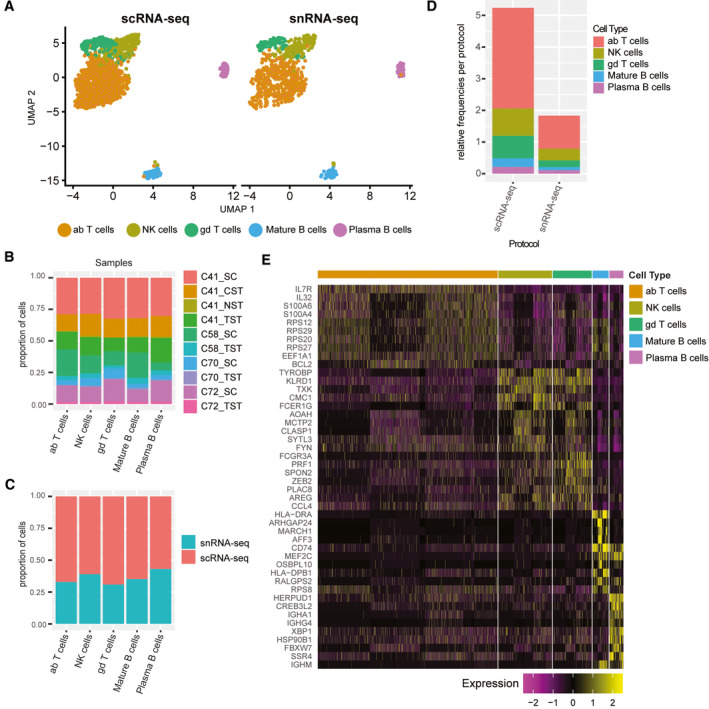
The critical functions of the human liver are coordinated through the interactions of hepatic parenchymal and non-parenchymal cells. Recent advances in single-cell transcriptional approaches have enabled an examination of the human liver with unprecedented resolution. However, dissociation-related cell perturbation can limit the ability to fully capture the human liver's parenchymal cell fraction, which limits the ability to comprehensively profile this organ. Here, we report the transcriptional landscape of 73,295 cells from the human liver using matched single-cell RNA sequencing (scRNA-seq) and single-nucleus RNA sequencing (snRNA-seq). The addition of snRNA-seq enabled the characterization of interzonal hepatocytes at a single-cell resolution, revealed the presence of rare subtypes of liver mesenchymal cells, and facilitated the detection of cholangiocyte progenitors that had only been observed during in vitro differentiation experiments. However, T and B lymphocytes and natural killer cells were only distinguishable using scRNA-seq, highlighting the importance of applying both technologies to obtain a complete map of tissue-resident cell types. We validated the distinct spatial distribution of the hepatocyte, cholangiocyte, and mesenchymal cell populations by an independent spatial transcriptomics data set and immunohistochemistry. Conclusion: Our study provides a systematic comparison of the transcriptomes captured by scRNA-seq and snRNA-seq and delivers a high-resolution map of the parenchymal cell populations in the healthy human liver.
© 2021 The Authors. Hepatology Communications published by Wiley Periodicals LLC on behalf of American Association for the Study of Liver Diseases.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
