

Idéfix: identifying accidental sample mix-ups in biobanks using polygenic scores
- PMID: 34792549
- PMCID: PMC8796367
- DOI: 10.1093/bioinformatics/btab783
Idéfix: identifying accidental sample mix-ups in biobanks using polygenic scores
Abstract
Motivation: Identifying sample mix-ups in biobanks is essential to allow the repurposing of genetic data for clinical pharmacogenetics. Pharmacogenetic advice based on the genetic information of another individual is potentially harmful. Existing methods for identifying mix-ups are limited to datasets in which additional omics data (e.g. gene expression) is available. Cohorts lacking such data can only use sex, which can reveal only half of the mix-ups. Here, we describe Idéfix, a method for the identification of accidental sample mix-ups in biobanks using polygenic scores.
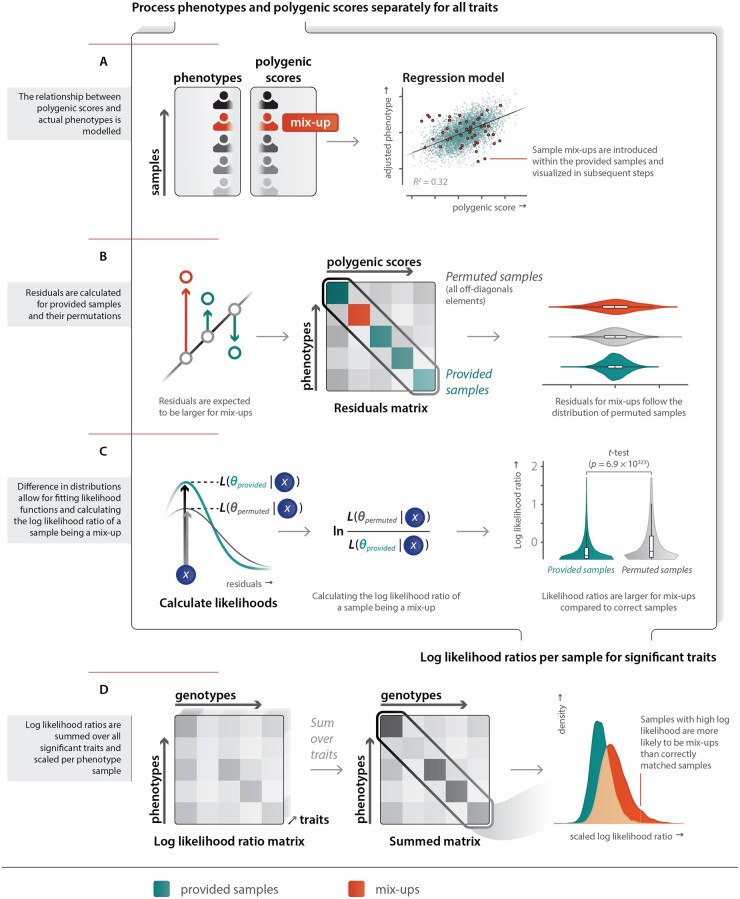
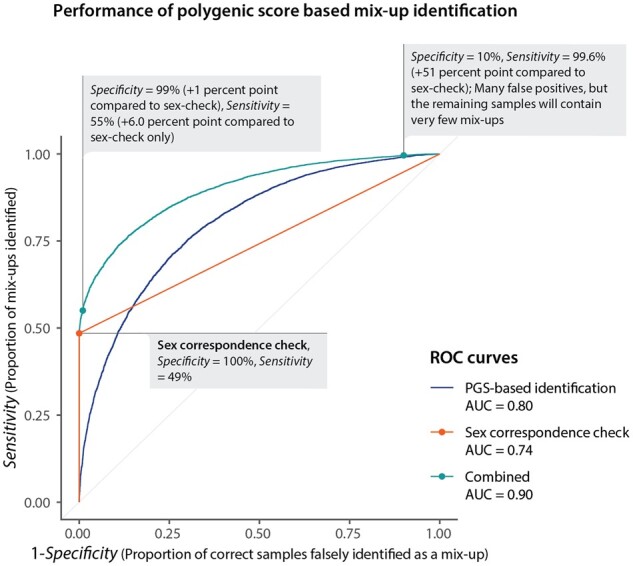
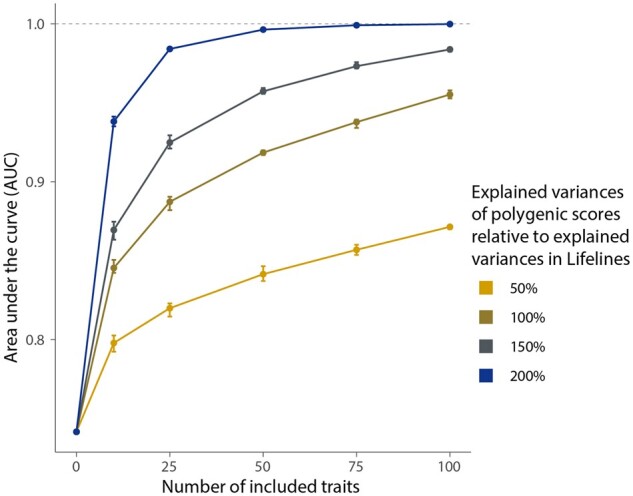
Results: In the Lifelines population-based biobank, we calculated polygenic scores (PGSs) for 25 traits for 32 786 participants. We then applied Idéfix to compare the actual phenotypes to PGSs, and to use the relative discordance that is expected for mix-ups, compared to correct samples. In a simulation, using induced mix-ups, Idéfix reaches an AUC of 0.90 using 25 polygenic scores and sex. This is a substantial improvement over using only sex, which has an AUC of 0.75. Subsequent simulations present Idéfix's potential in varying datasets with more powerful PGSs. This suggests its performance will likely improve when more highly powered GWASs for commonly measured traits will become available. Idéfix can be used to identify a set of high-quality participants for whom it is very unlikely that they reflect sample mix-ups, and for these participants we can use genetic data for clinical purposes, such as pharmacogenetic profiles. For instance, in Lifelines, we can select 34.4% of participants, reducing the sample mix-up rate from 0.15% to 0.01%.
Availabilityand implementation: Idéfix is freely available at https://github.com/molgenis/systemsgenetics/wiki/Idefix. The individual-level data that support the findings were obtained from the Lifelines biobank under project application number ov16_0365. Data is made available upon reasonable request submitted to the LifeLines Research office (research@lifelines.nl, https://www.lifelines.nl/researcher/how-to-apply/apply-here).
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press.
Figures
References
-
- Buyske S. et al. (2009) When a case is not a case: effects of phenotype misclassification on power and sample size requirements for the transmission disequilibrium test with affected child trios. Hum. Hered., 67, 287–292. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
