

KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression
- PMID: 34793333
- PMCID: PMC8803339
- DOI: 10.1172/JCI147191
KLF2 regulates neutrophil activation and thrombosis in cardiac hypertrophy and heart failure progression
Abstract
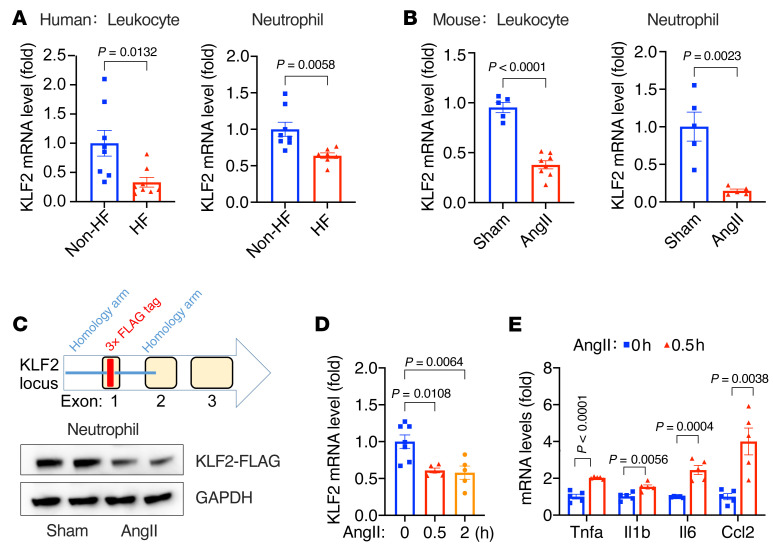
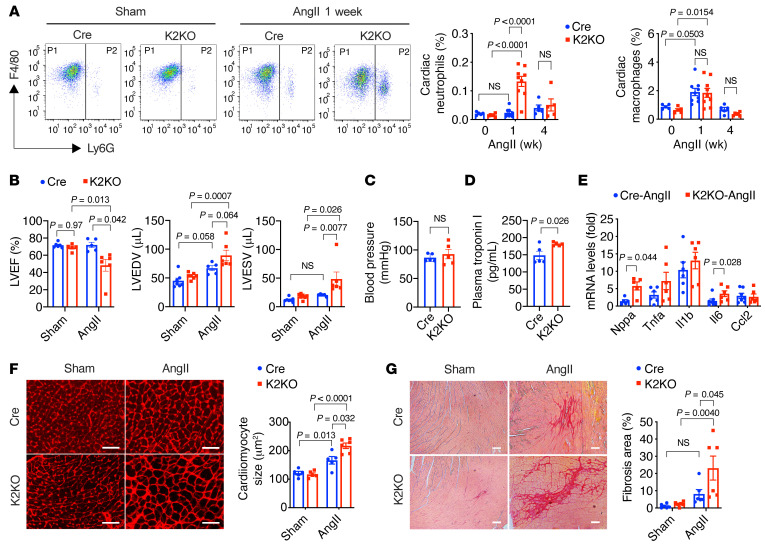
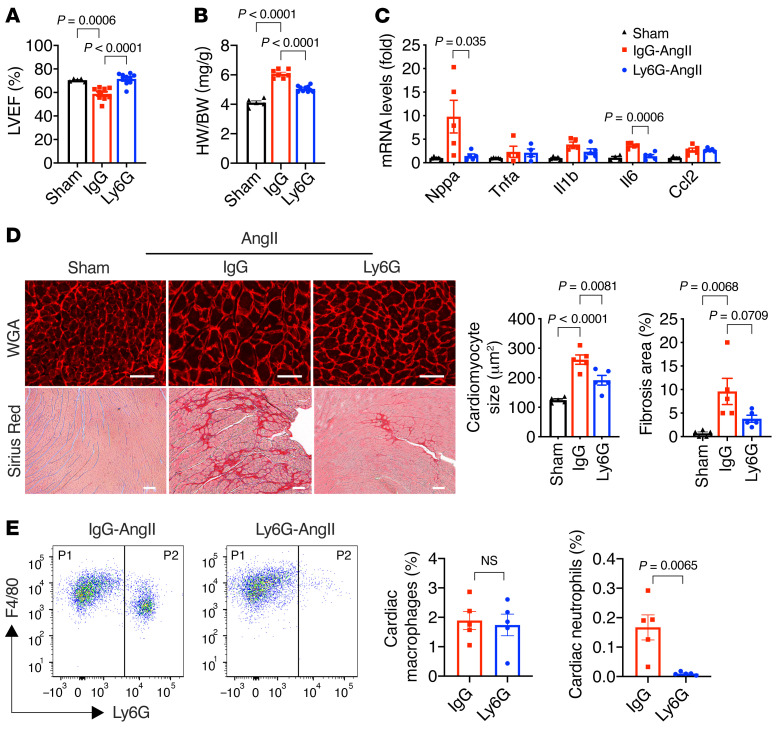
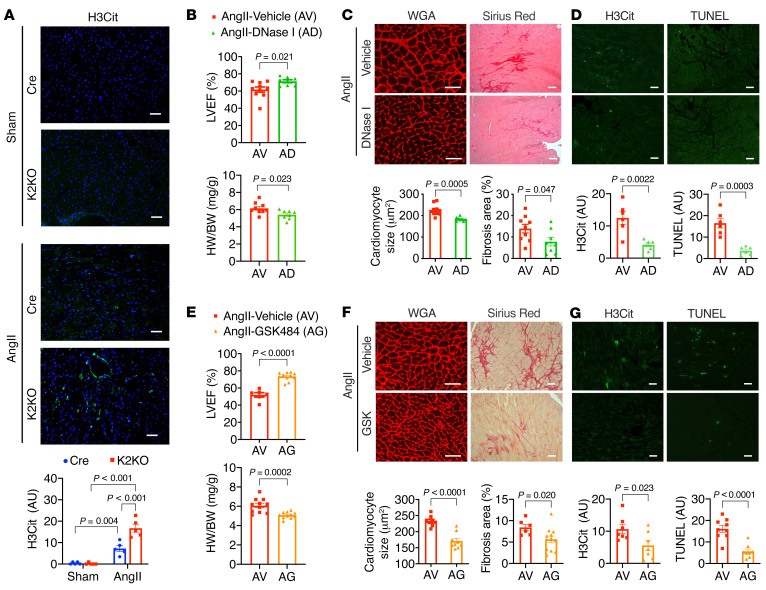
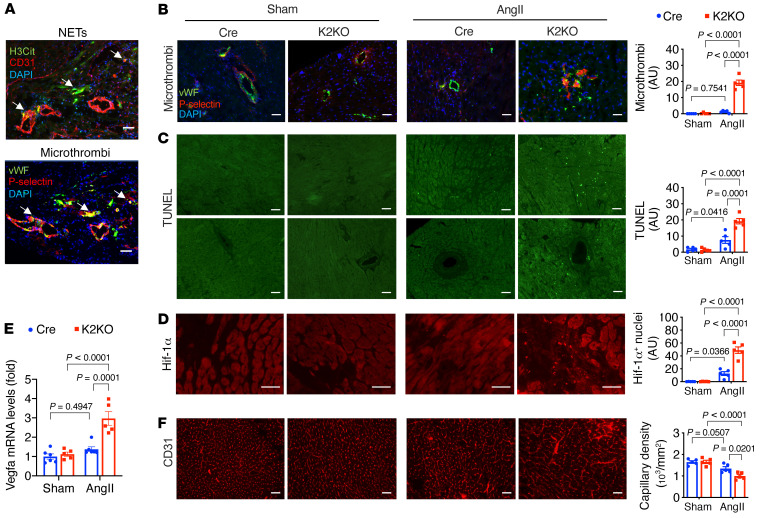
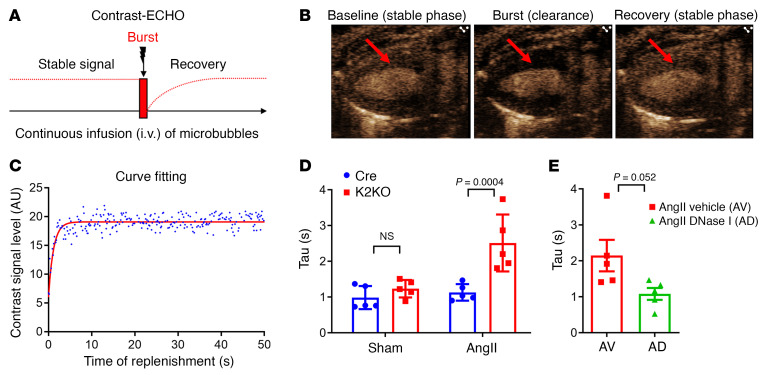
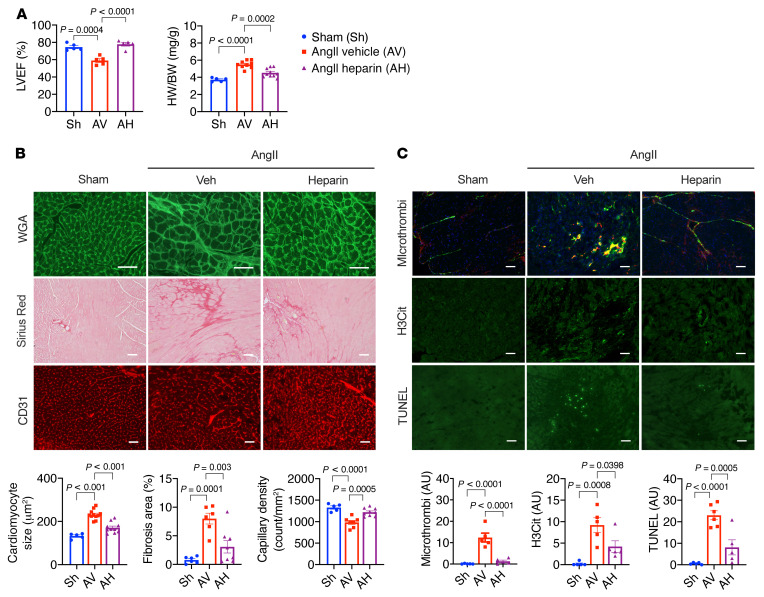
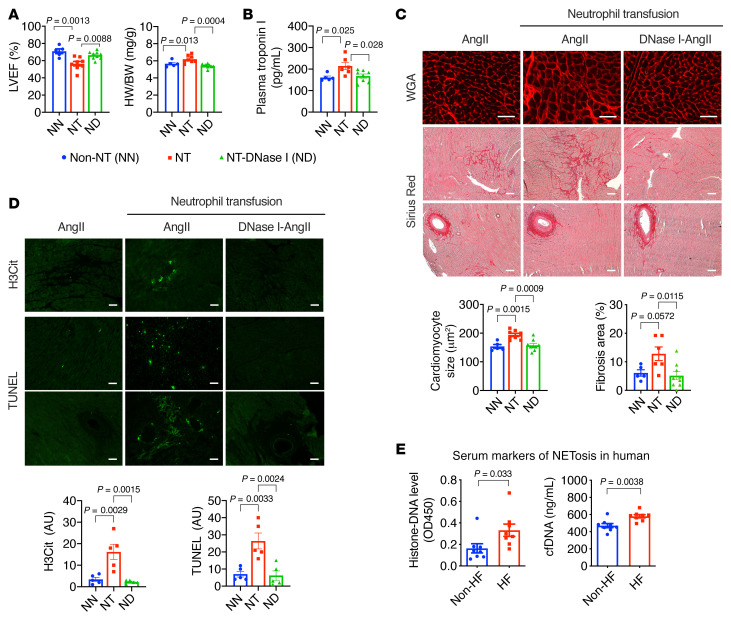
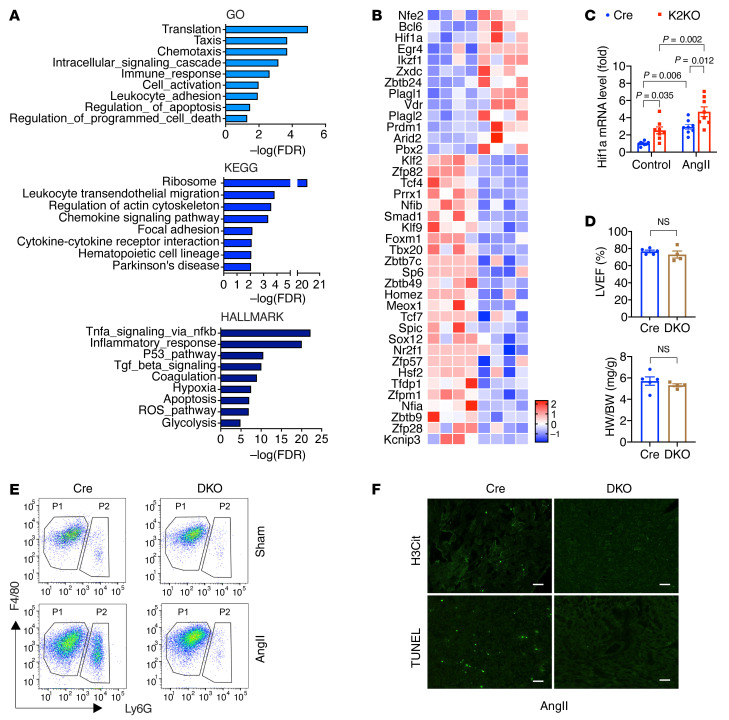
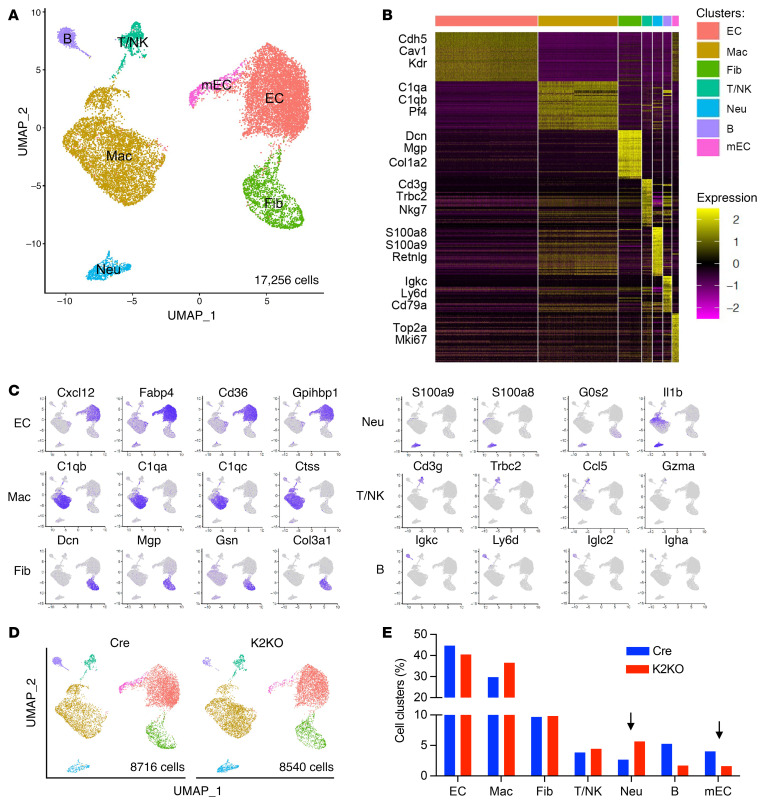
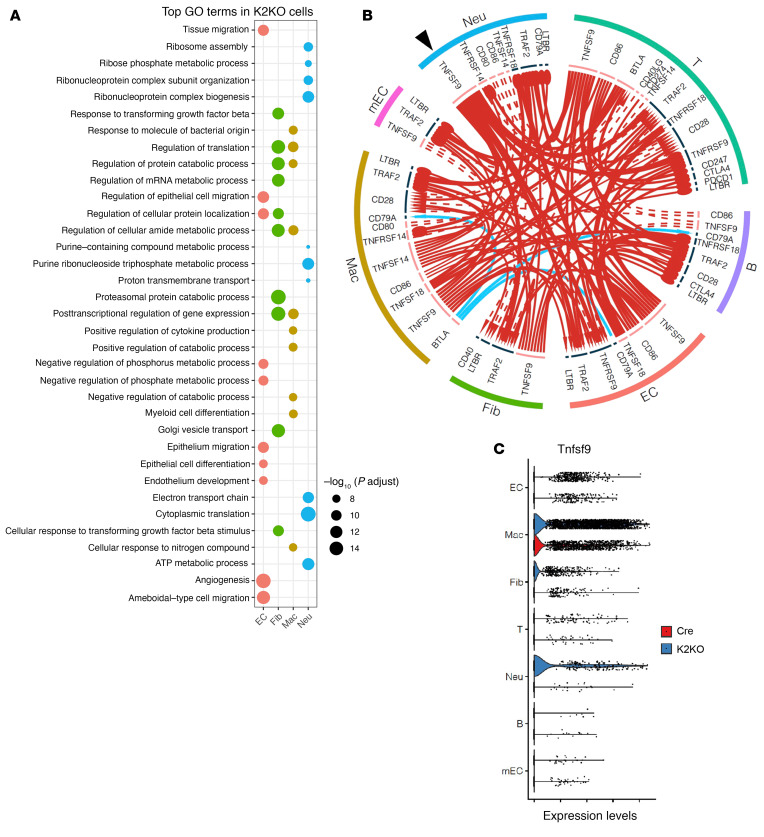
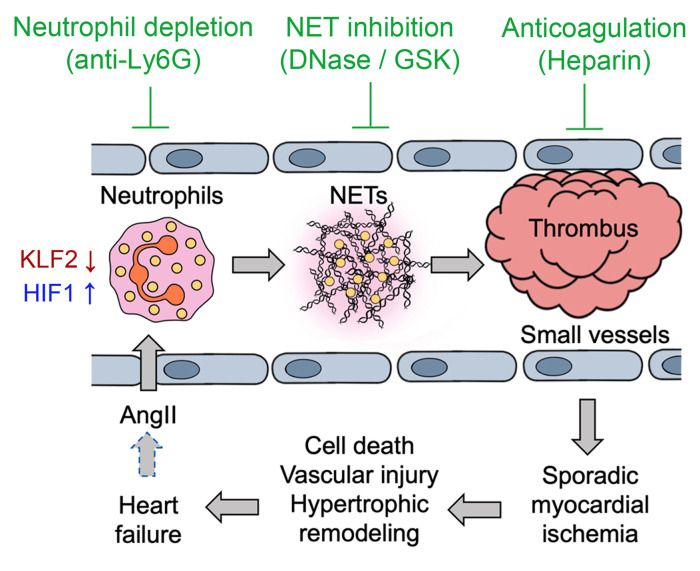
It is widely recognized that inflammation plays a critical role in cardiac hypertrophy and heart failure. However, clinical trials targeting cytokines have shown equivocal effects, indicating the need for a deeper understanding of the precise role of inflammation and inflammatory cells in heart failure. Leukocytes from human subjects and a rodent model of heart failure were characterized by a marked reduction in expression of Klf2 mRNA. Using a mouse model of angiotensin II-induced nonischemic cardiac dysfunction, we showed that neutrophils played an essential role in the pathogenesis and progression of heart failure. Mechanistically, chronic angiotensin II infusion activated a neutrophil KLF2/NETosis pathway that triggered sporadic thrombosis in small myocardial vessels, leading to myocardial hypoxia, cell death, and hypertrophy. Conversely, targeting neutrophils, neutrophil extracellular traps (NETs), or thrombosis ameliorated these pathological changes and preserved cardiac dysfunction. KLF2 regulated neutrophil activation in response to angiotensin II at the molecular level, partly through crosstalk with HIF1 signaling. Taken together, our data implicate neutrophil-mediated immunothrombotic dysregulation as a critical pathogenic mechanism leading to cardiac hypertrophy and heart failure. This neutrophil KLF2-NETosis-thrombosis mechanism underlying chronic heart failure can be exploited for therapeutic gain by therapies targeting neutrophils, NETosis, or thrombosis.
Keywords: Cardiology; Cardiovascular disease; Inflammation; Neutrophils; Transcription.
Conflict of interest statement
Figures
Comment in
-
Neutrophil extracellular traps in cardiac hypertrophy: a KLF2 perspective.J Clin Invest. 2022 Feb 1;132(3):e156453. doi: 10.1172/JCI156453. J Clin Invest. 2022. PMID: 35104806 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
