

Closing the gap: Systematic integration of multiplexed functional data resolves variants of uncertain significance in BRCA1, TP53, and PTEN
- PMID: 34793697
- PMCID: PMC8715144
- DOI: 10.1016/j.ajhg.2021.11.001
Closing the gap: Systematic integration of multiplexed functional data resolves variants of uncertain significance in BRCA1, TP53, and PTEN
Abstract
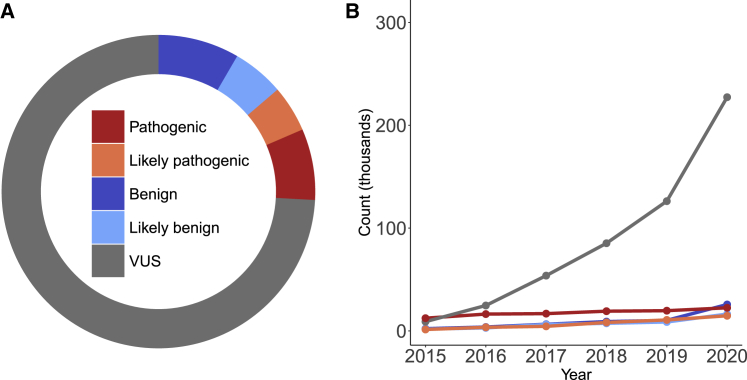
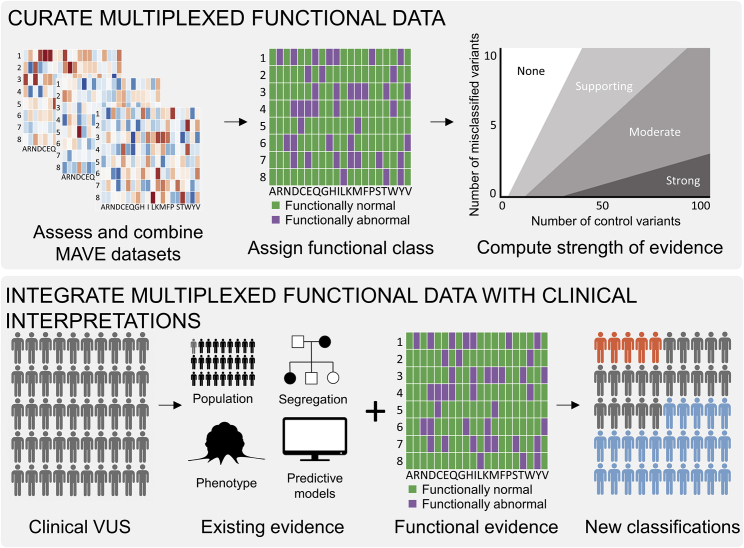
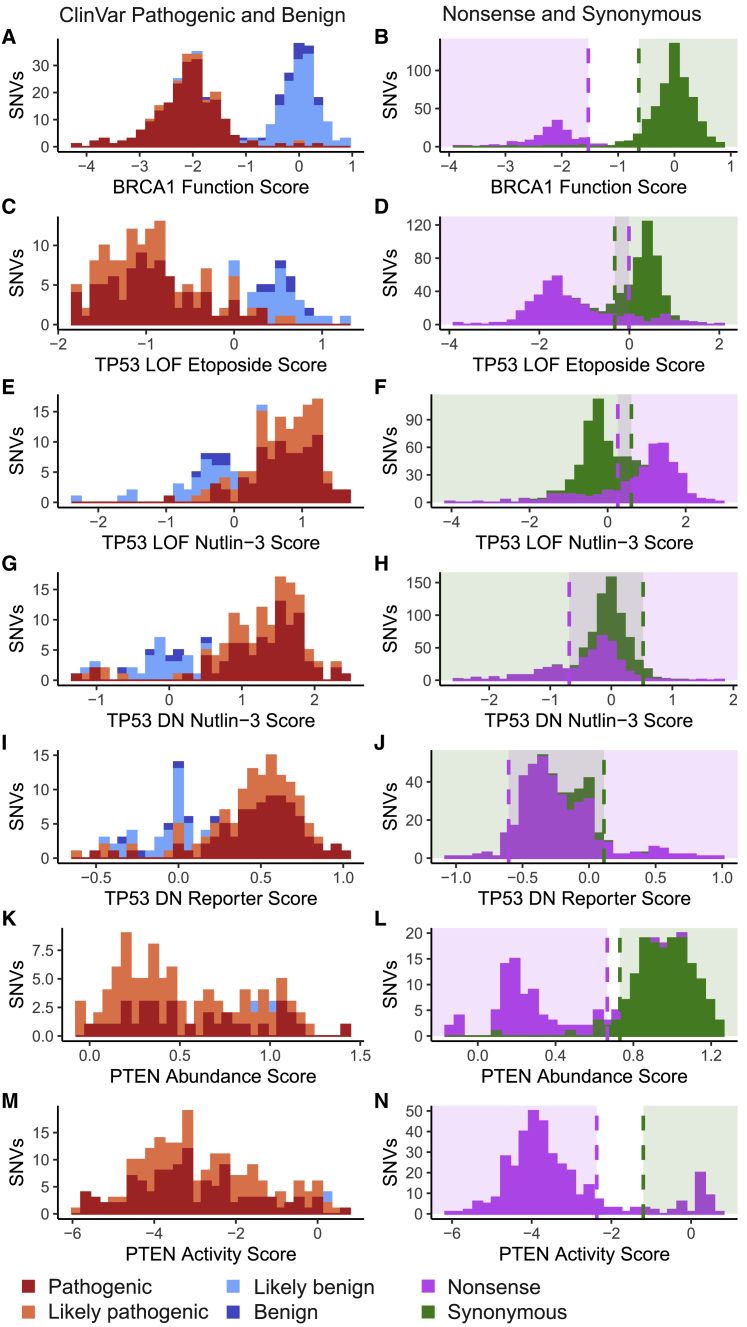
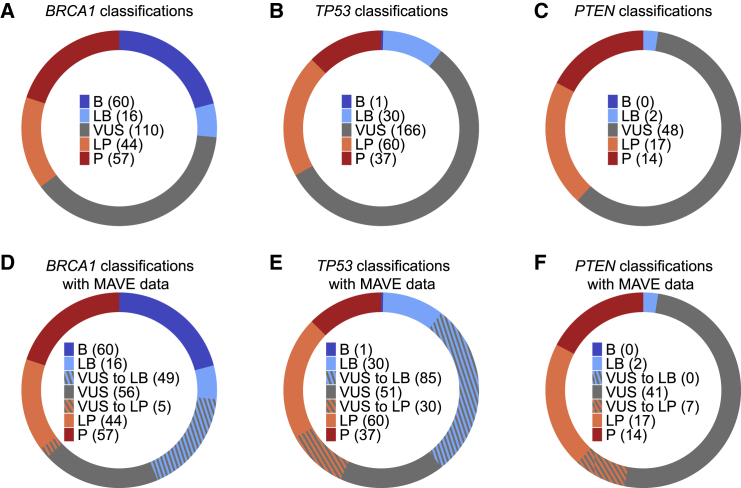
Clinical interpretation of missense variants is challenging because the majority identified by genetic testing are rare and their functional effects are unknown. Consequently, most variants are of uncertain significance and cannot be used for clinical diagnosis or management. Although not much can be done to ameliorate variant rarity, multiplexed assays of variant effect (MAVEs), where thousands of single-nucleotide variant effects are simultaneously measured experimentally, provide functional evidence that can help resolve variants of unknown significance (VUSs). However, a rigorous assessment of the clinical value of multiplexed functional data for variant interpretation is lacking. Thus, we systematically combined previously published BRCA1, TP53, and PTEN multiplexed functional data with phenotype and family history data for 324 VUSs identified by a single diagnostic testing laboratory. We curated 49,281 variant functional scores from MAVEs for these three genes and integrated four different TP53 multiplexed functional datasets into a single functional prediction for each variant by using machine learning. We then determined the strength of evidence provided by each multiplexed functional dataset and reevaluated 324 VUSs. Multiplexed functional data were effective in driving variant reclassification when combined with clinical data, eliminating 49% of VUSs for BRCA1, 69% for TP53, and 15% for PTEN. Thus, multiplexed functional data, which are being generated for numerous genes, are poised to have a major impact on clinical variant interpretation.
Keywords: BRCA1; MAVE; PTEN; TP53; functional data; variant interpretation.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.N.D. is an employee of Adaptive. M.E.R., K.M., F.H., T.P., and R.K. are employees of Ambry Genetics. The remaining authors declare no competing interests.
Figures
References
-
- Maxwell K.N., Hart S.N., Vijai J., Schrader K.A., Slavin T.P., Thomas T., Wubbenhorst B., Ravichandran V., Moore R.M., Hu C., et al. Evaluation of ACMG-Guideline-Based Variant Classification of Cancer Susceptibility and Non-Cancer-Associated Genes in Families Affected by Breast Cancer. Am. J. Hum. Genet. 2016;98:801–817. - PMC - PubMed
-
- LaDuca H., Polley E.C., Yussuf A., Hoang L., Gutierrez S., Hart S.N., Yadav S., Hu C., Na J., Goldgar D.E., et al. A clinical guide to hereditary cancer panel testing: evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet. Med. 2020;22:407–415. - PMC - PubMed
-
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
