

Single-cell analysis of diverse immune phenotypes in malignant pleural effusion
- PMID: 34795282
- PMCID: PMC8602344
- DOI: 10.1038/s41467-021-27026-9
Single-cell analysis of diverse immune phenotypes in malignant pleural effusion
Abstract
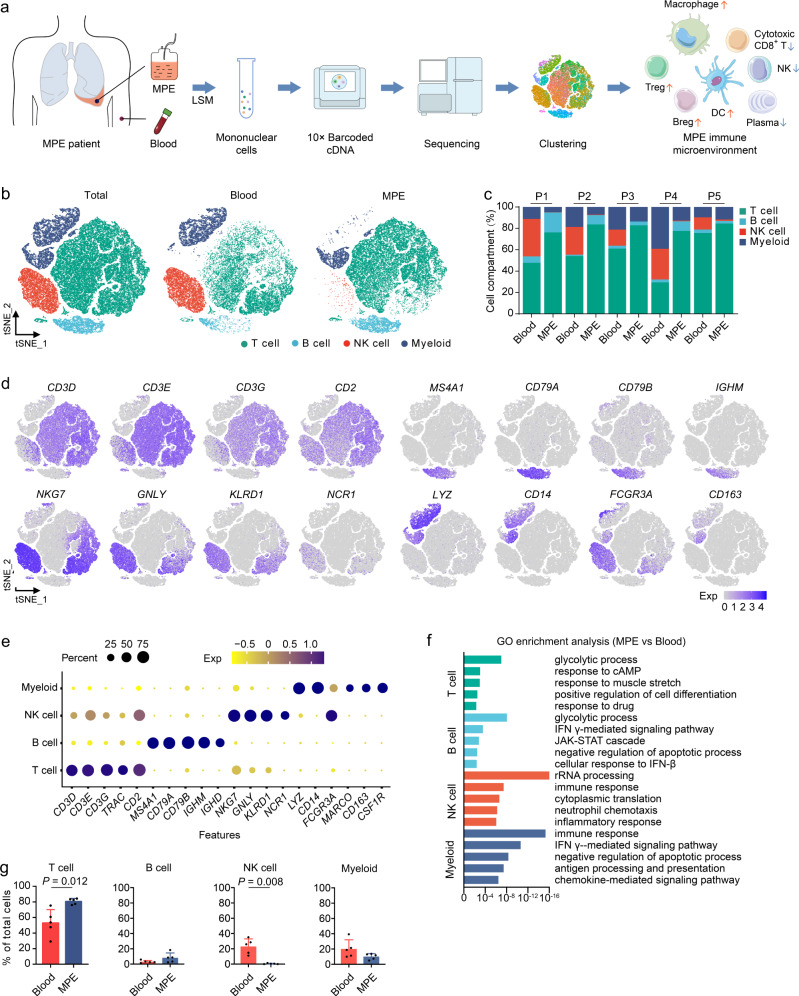
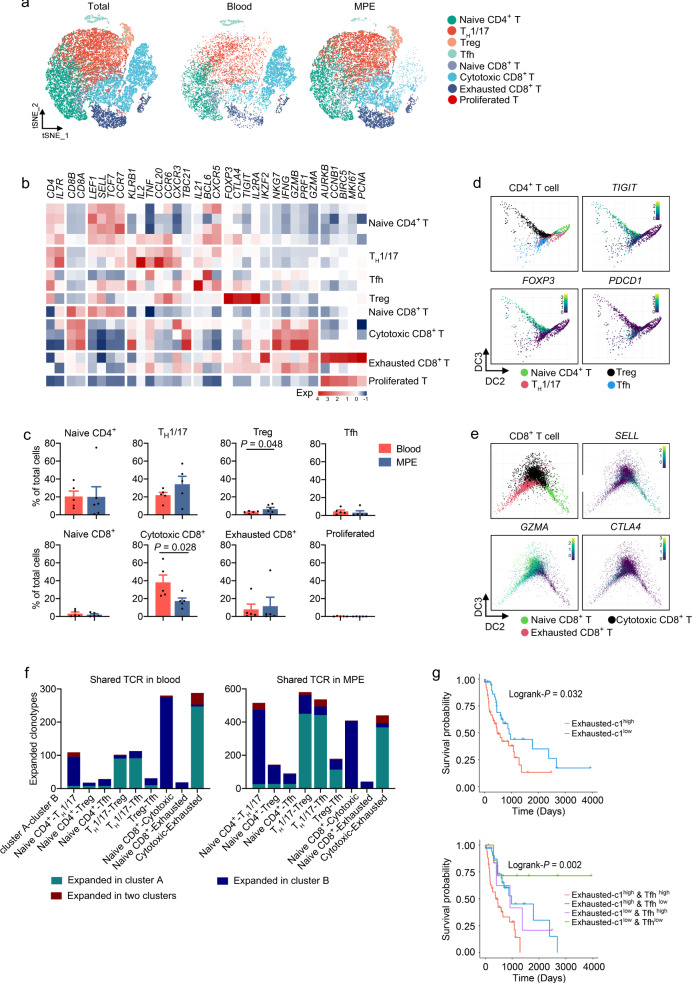
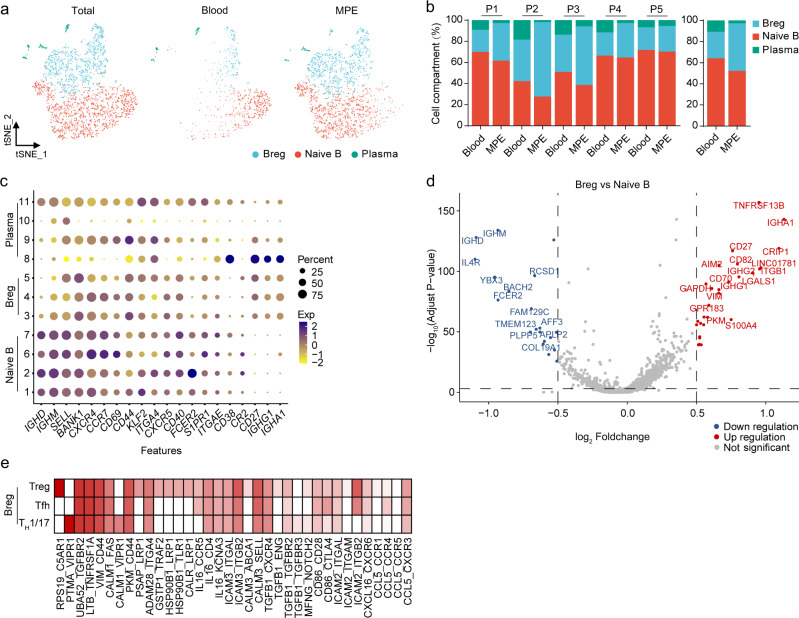
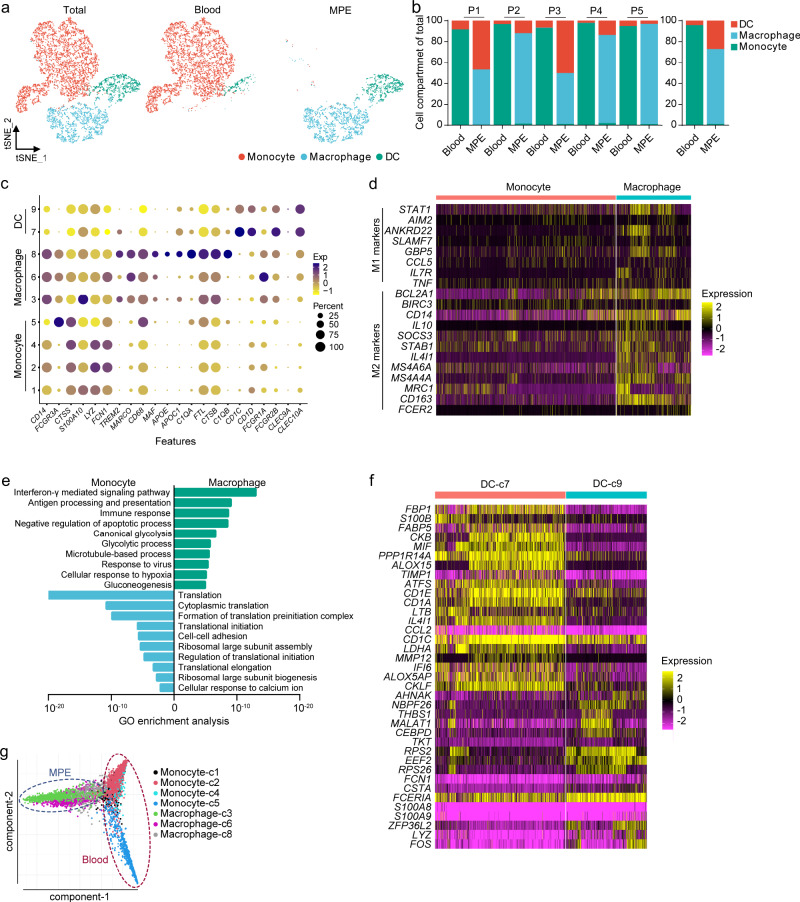
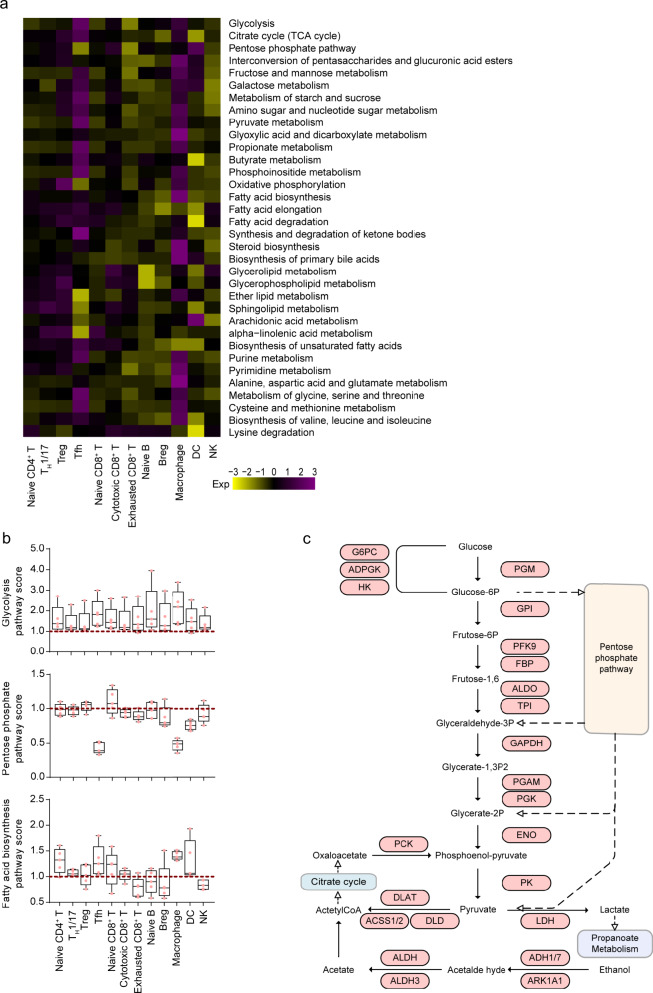
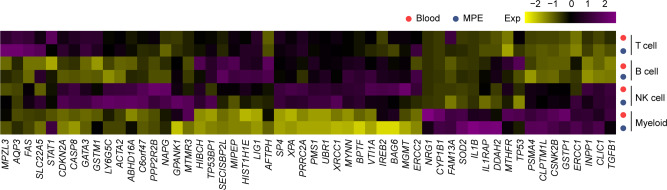
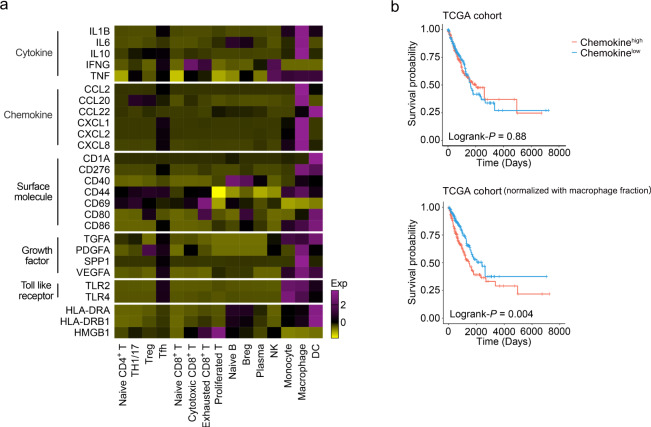
The complex interactions among different immune cells have important functions in the development of malignant pleural effusion (MPE). Here we perform single-cell RNA sequencing on 62,382 cells from MPE patients induced by non-small cell lung cancer to describe the composition, lineage, and functional states of infiltrating immune cells in MPE. Immune cells in MPE display a number of transcriptional signatures enriched for regulatory T cells, B cells, macrophages, and dendritic cells compared to corresponding counterparts in blood. Helper T, cytotoxic T, regulatory T, and T follicular helper cells express multiple immune checkpoints or costimulatory molecules. Cell-cell interaction analysis identifies regulatory B cells with more interactions with CD4+ T cells compared to CD8+ T cells. Macrophages are transcriptionally heterogeneous and conform to M2 polarization characteristics. In addition, immune cells in MPE show the general up-regulation of glycolytic pathways associated with the hypoxic microenvironment. These findings show a detailed atlas of immune cells in human MPE and enhance the understanding of potential diagnostic and therapeutic targets in advanced non-small cell lung cancer.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Taghizadeh N, Fortin M, Tremblay A. US hospitalizations for malignant pleural effusions: data from the 2012 national inpatient sample. Chest. 2017;151:845–854. - PubMed
-
- Walker S, Mercer R, Maskell N, Rahman NM. Malignant pleural effusion management: keeping the flood gates shut. Lancet Respir. Med. 2020;8:609–618. - PubMed
-
- Ryu JS, et al. Prognostic impact of minimal pleural effusion in non-small-cell lung cancer. J. Clin. Oncol. 2014;32:960–967. - PubMed
-
- Bibby, A. C. et al. ERS/EACTS statement on the management of malignant pleural effusions. Eur. Respir. J.52, 1800349 (2018). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
