

The Epigenome in Neurodevelopmental Disorders
- PMID: 34803599
- PMCID: PMC8595945
- DOI: 10.3389/fnins.2021.776809
The Epigenome in Neurodevelopmental Disorders
Abstract
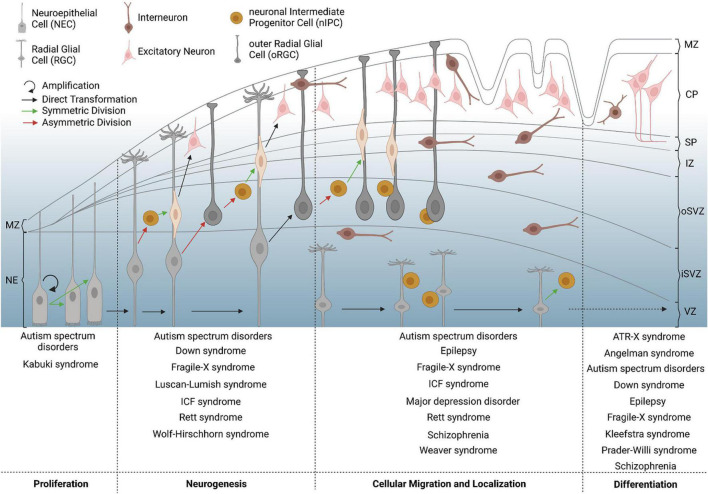
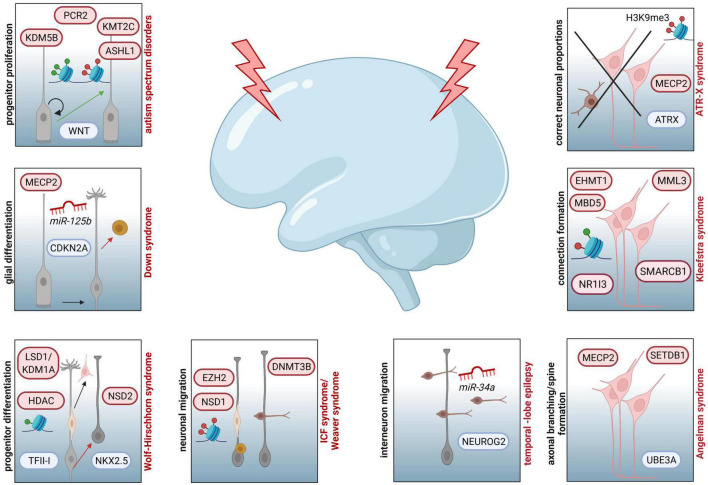
Neurodevelopmental diseases (NDDs), such as autism spectrum disorders, epilepsy, and schizophrenia, are characterized by diverse facets of neurological and psychiatric symptoms, differing in etiology, onset and severity. Such symptoms include mental delay, cognitive and language impairments, or restrictions to adaptive and social behavior. Nevertheless, all have in common that critical milestones of brain development are disrupted, leading to functional deficits of the central nervous system and clinical manifestation in child- or adulthood. To approach how the different development-associated neuropathologies can occur and which risk factors or critical processes are involved in provoking higher susceptibility for such diseases, a detailed understanding of the mechanisms underlying proper brain formation is required. NDDs rely on deficits in neuronal identity, proportion or function, whereby a defective development of the cerebral cortex, the seat of higher cognitive functions, is implicated in numerous disorders. Such deficits can be provoked by genetic and environmental factors during corticogenesis. Thereby, epigenetic mechanisms can act as an interface between external stimuli and the genome, since they are known to be responsive to external stimuli also in cortical neurons. In line with that, DNA methylation, histone modifications/variants, ATP-dependent chromatin remodeling, as well as regulatory non-coding RNAs regulate diverse aspects of neuronal development, and alterations in epigenomic marks have been associated with NDDs of varying phenotypes. Here, we provide an overview of essential steps of mammalian corticogenesis, and discuss the role of epigenetic mechanisms assumed to contribute to pathophysiological aspects of NDDs, when being disrupted.
Keywords: DNA methylation; chromatin remodeling; corticogenesis; epigenetics; histone modification; neuropsychiatry; non-coding RNAs.
Copyright © 2021 Reichard and Zimmer-Bensch.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources