

Genetic recombination in fast-spreading coxsackievirus A6 variants: a potential role in evolution and pathogenicity
- PMID: 34804589
- PMCID: PMC8597624
- DOI: 10.1093/ve/veaa048
Genetic recombination in fast-spreading coxsackievirus A6 variants: a potential role in evolution and pathogenicity
Abstract
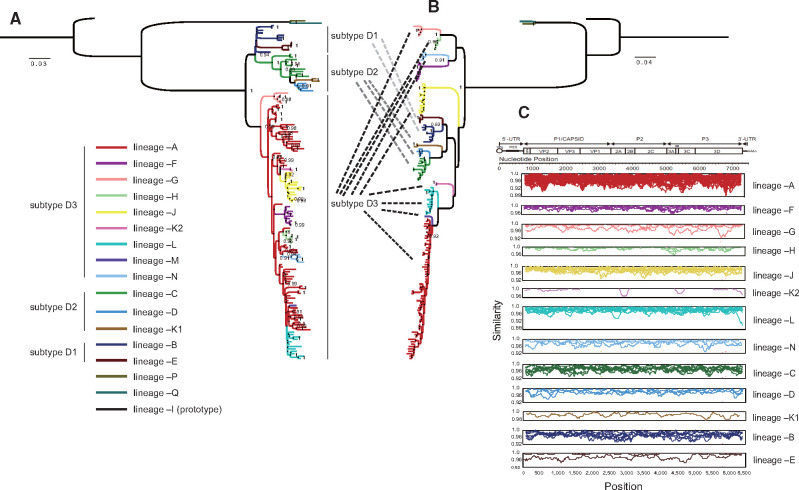
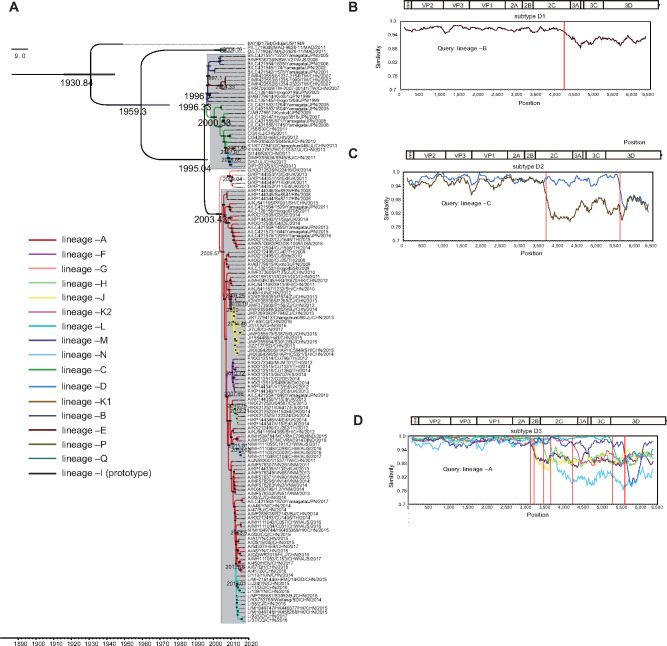
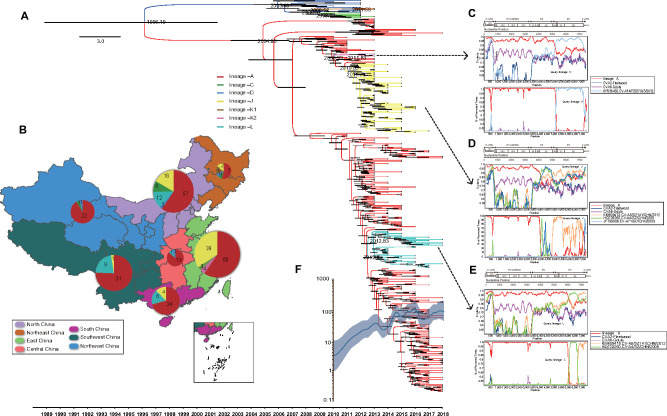
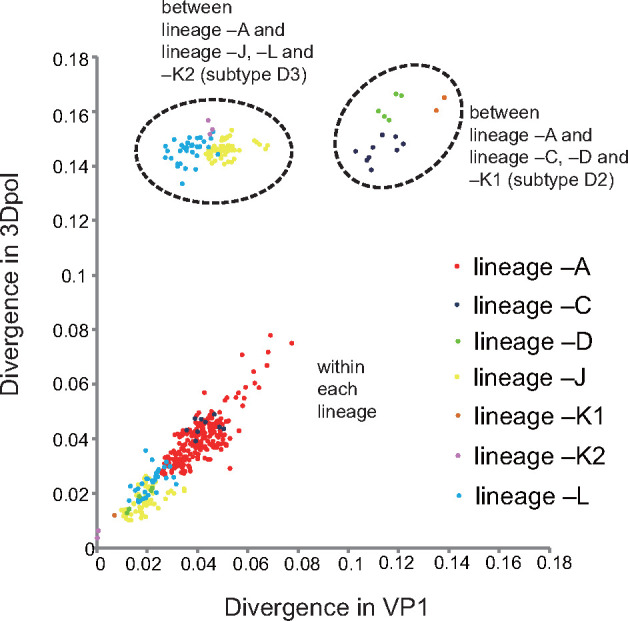
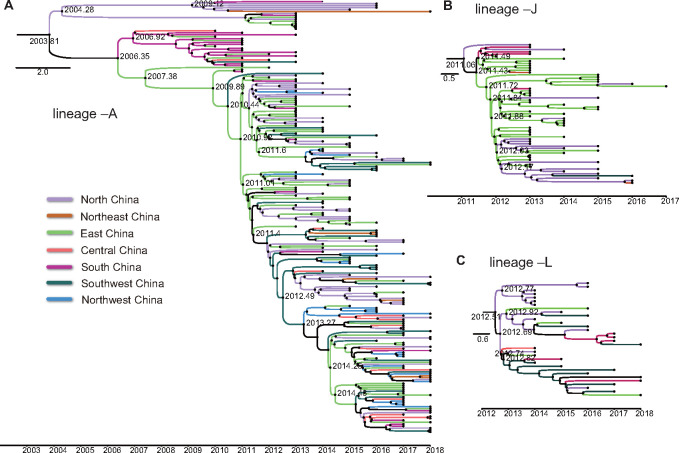
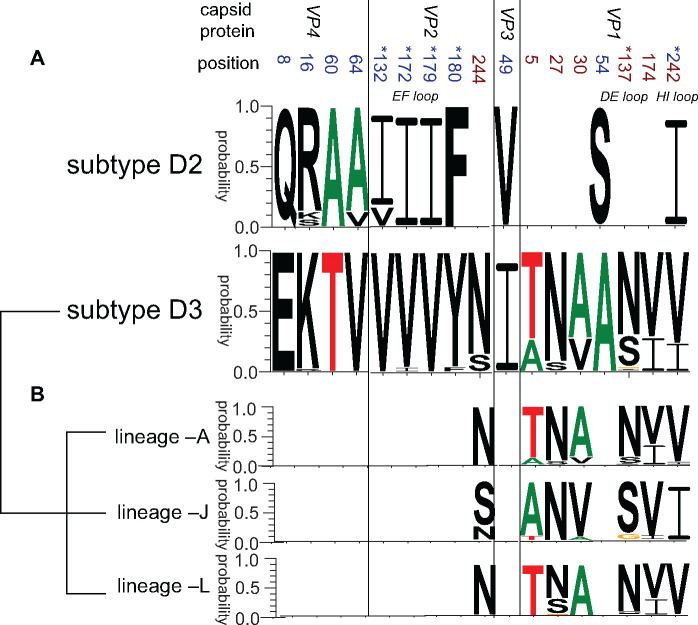
Hand, foot, and mouth disease (HFMD) is a common global epidemic. From 2008 onwards, many HFMD outbreaks caused by coxsackievirus A6 (CV-A6) have been reported worldwide. Since 2013, with a dramatically increasing number of CV-A6-related HFMD cases, CV-A6 has become the predominant HFMD pathogen in mainland China. Phylogenetic analysis based on the VP1 capsid gene revealed that subtype D3 dominated the CV-A6 outbreaks. Here, we performed a large-scale (near) full-length genetic analysis of global and Chinese CV-A6 variants, including 158 newly sequenced samples collected extensively in mainland China between 2010 and 2018. During the global transmission of subtype D3 of CV-A6, the noncapsid gene continued recombining, giving rise to a series of viable recombinant hybrids designated evolutionary lineages, and each lineage displayed internal consistency in both genetic and epidemiological features. The emergence of lineage -A since 2005 has triggered CV-A6 outbreaks worldwide, with a rate of evolution estimated at 4.17 × 10-3 substitutions site- 1 year-1 based on a large number of monophyletic open reading frame sequences, and created a series of lineages chronologically through varied noncapsid recombination events. In mainland China, lineage -A has generated another two novel widespread lineages (-J and -L) through recombination within the enterovirus A gene pool, with robust estimates of occurrence time. Lineage -A, -J, and -L infections presented dissimilar clinical manifestations, indicating that the conservation of the CV-A6 capsid gene resulted in high transmissibility, but the lineage-specific noncapsid gene might influence pathogenicity. Potentially important amino acid substitutions were further predicted among CV-A6 variants. The evolutionary phenomenon of noncapsid polymorphism within the same subtype observed in CV-A6 was uncommon in other leading HFMD pathogens; such frequent recombination happened in fast-spreading CV-A6, indicating that the recovery of deleterious genomes may still be ongoing within CV-A6 quasispecies. CV-A6-related HFMD outbreaks have caused a significant public health burden and pose a great threat to children's health; therefore, further surveillance is greatly needed to understand the full genetic diversity of CV-A6 in mainland China.
Keywords: Coxsackievirus A6; evolutionary dynamics; foot and mouth disease; genetic recombination; hand; pathogenicity; phylogeny; whole-genome analysis.
© The Author(s) 2020. Published by Oxford University Press.
Figures
Similar articles
-
Molecular Evolutionary Dynamics of Coxsackievirus A6 Causing Hand, Foot, and Mouth Disease From 2021 to 2023 in China: Genomic Epidemiology Study.JMIR Public Health Surveill. 2024 Jul 31;10:e59604. doi: 10.2196/59604. JMIR Public Health Surveill. 2024. PMID: 39087568 Free PMC article.
-
Emergence, circulation, and spatiotemporal phylogenetic analysis of coxsackievirus a6- and coxsackievirus a10-associated hand, foot, and mouth disease infections from 2008 to 2012 in Shenzhen, China.J Clin Microbiol. 2013 Nov;51(11):3560-6. doi: 10.1128/JCM.01231-13. Epub 2013 Aug 21. J Clin Microbiol. 2013. PMID: 23966496 Free PMC article.
-
Divergent Pathogenic Properties of Circulating Coxsackievirus A6 Associated with Emerging Hand, Foot, and Mouth Disease.J Virol. 2018 May 14;92(11):e00303-18. doi: 10.1128/JVI.00303-18. Print 2018 Jun 1. J Virol. 2018. PMID: 29563294 Free PMC article.
-
A review of the recombination events, mechanisms and consequences of Coxsackievirus A6.Infect Med (Beijing). 2024 May 1;3(2):100115. doi: 10.1016/j.imj.2024.100115. eCollection 2024 Jun. Infect Med (Beijing). 2024. PMID: 38974347 Free PMC article. Review.
-
A review and meta-analysis of the epidemiology and clinical presentation of coxsackievirus A6 causing hand-foot-mouth disease in China and global implications.Rev Med Virol. 2020 Mar;30(2):e2087. doi: 10.1002/rmv.2087. Epub 2019 Dec 6. Rev Med Virol. 2020. PMID: 31811676 Review.
Cited by
-
Coxsackievirus A6 Recombinant Subclades D3/A and D3/H Were Predominant in Hand-Foot-And-Mouth Disease Outbreaks in the Paediatric Population, France, 2010-2018.Viruses. 2022 May 17;14(5):1078. doi: 10.3390/v14051078. Viruses. 2022. PMID: 35632819 Free PMC article.
-
Multiple molecular characteristics of circulating enterovirus types among pediatric hand, foot and mouth disease patients after EV71 vaccination campaign in Wuxi, China.Epidemiol Infect. 2022 Apr 27;150:1-19. doi: 10.1017/S0950268822000784. Online ahead of print. Epidemiol Infect. 2022. PMID: 35473720 Free PMC article.
-
Transition of D3c branch and novel recombination events contribute to the diversity of Coxsackievirus A6 in Beijing, China, from 2019 to 2023.Virus Evol. 2025 May 11;11(1):veaf036. doi: 10.1093/ve/veaf036. eCollection 2025. Virus Evol. 2025. PMID: 40574752 Free PMC article.
-
Coxsackievirus A6 Infection Causes Neurogenic Pathogenesis in a Neonatal Murine Model.Viruses. 2023 Feb 12;15(2):511. doi: 10.3390/v15020511. Viruses. 2023. PMID: 36851724 Free PMC article.
-
Epidemiological, etiological, and serological characteristics of hand, foot, and mouth disease in Guizhou Province, Southwest China, from 2008 to 2023.PLoS Negl Trop Dis. 2025 Aug 18;19(8):e0013394. doi: 10.1371/journal.pntd.0013394. eCollection 2025 Aug. PLoS Negl Trop Dis. 2025. PMID: 40825065 Free PMC article.
References
-
- Blomberg J. et al. (1974) ‘Letter: New Enterovirus Type Associated with Epidemic of Aseptic Meningitis and-or Hand, Foot, and Mouth Disease’, The Lancet, 304: 112. - PubMed
LinkOut - more resources
Full Text Sources
