

Mitotic Errors Promote Genomic Instability and Leukemia in a Novel Mouse Model of Fanconi Anemia
- PMID: 34804941
- PMCID: PMC8602820
- DOI: 10.3389/fonc.2021.752933
Mitotic Errors Promote Genomic Instability and Leukemia in a Novel Mouse Model of Fanconi Anemia
Abstract
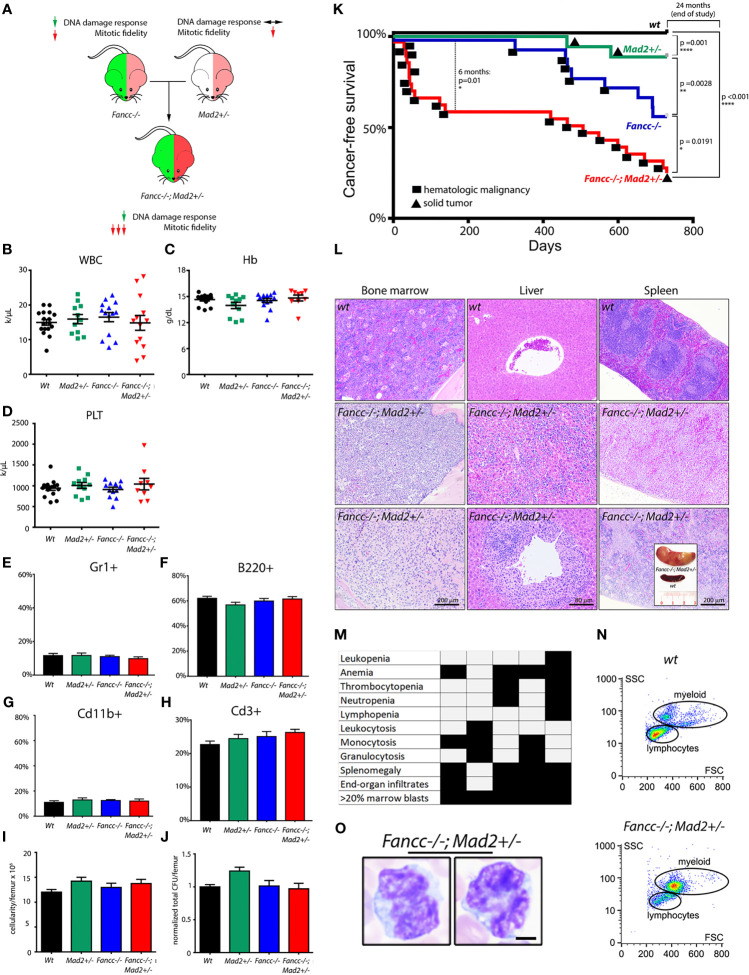
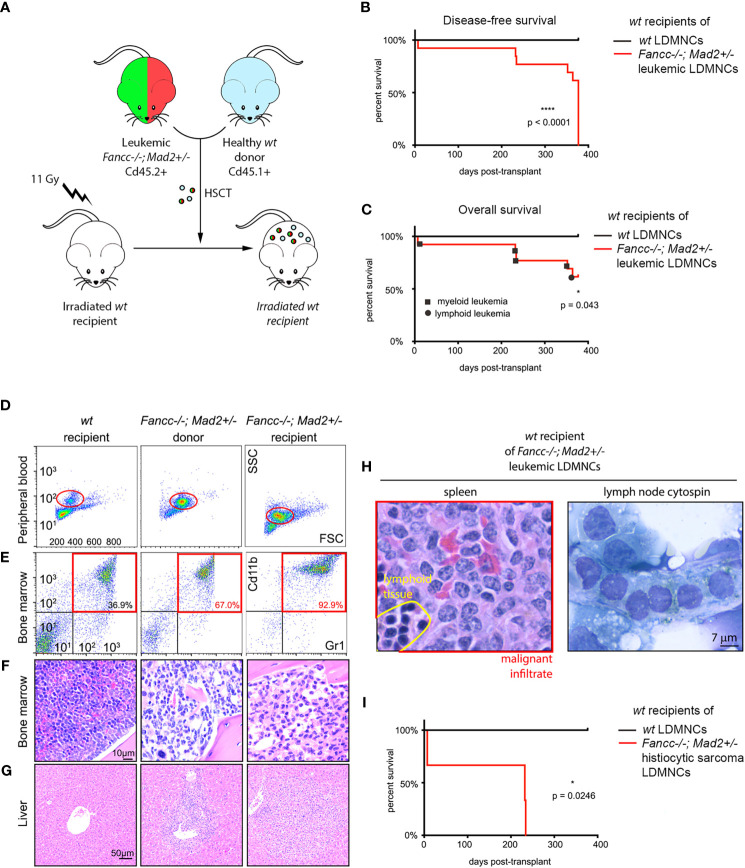
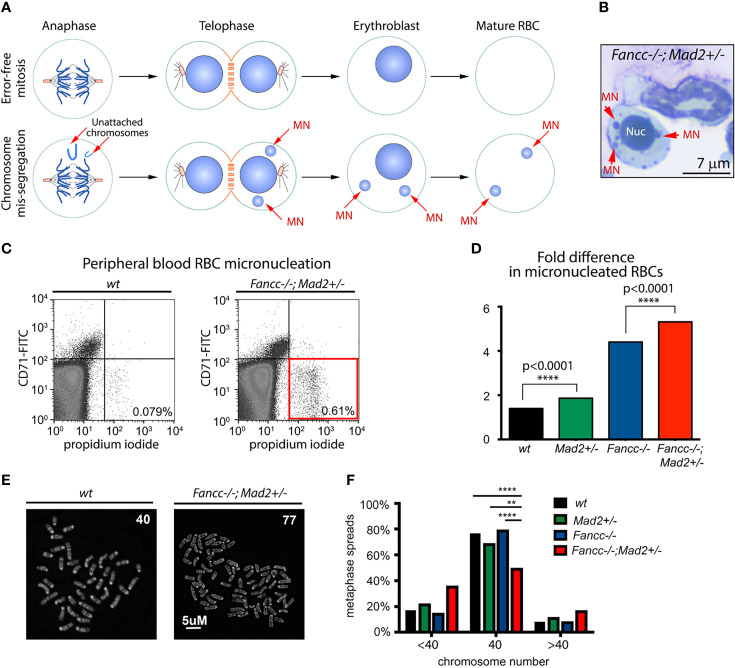
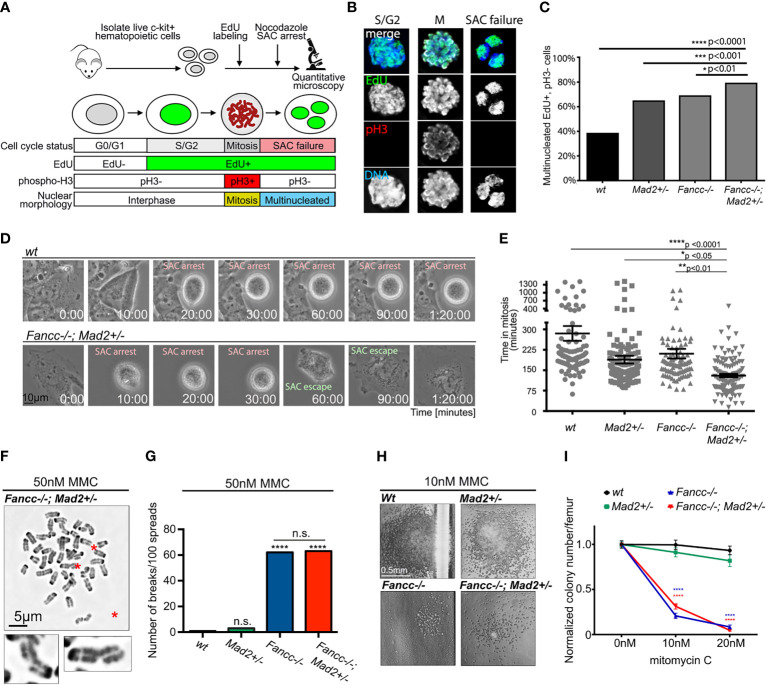
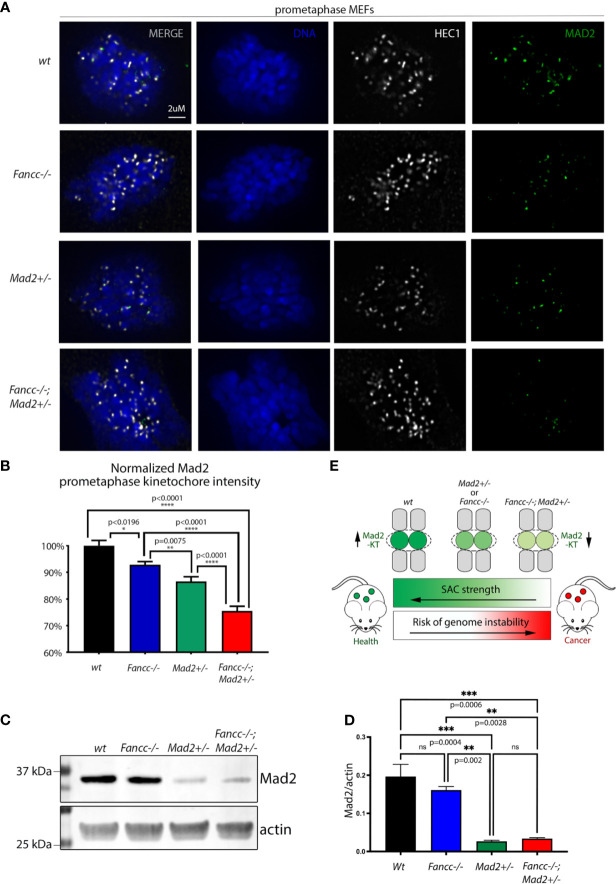
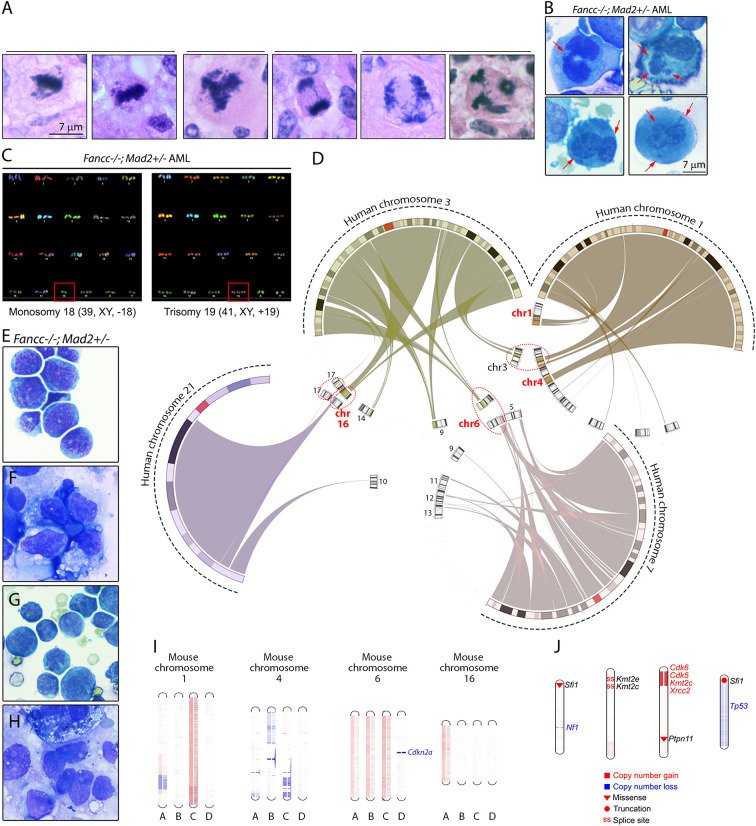
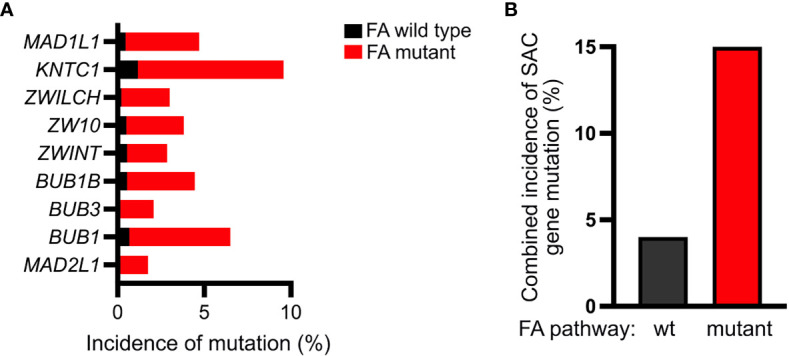
Fanconi anemia (FA) is a disease of genomic instability and cancer. In addition to DNA damage repair, FA pathway proteins are now known to be critical for maintaining faithful chromosome segregation during mitosis. While impaired DNA damage repair has been studied extensively in FA-associated carcinogenesis in vivo, the oncogenic contribution of mitotic abnormalities secondary to FA pathway deficiency remains incompletely understood. To examine the role of mitotic dysregulation in FA pathway deficient malignancies, we genetically exacerbated the baseline mitotic defect in Fancc-/- mice by introducing heterozygosity of the key spindle assembly checkpoint regulator Mad2. Fancc-/-;Mad2+/- mice were viable, but died from acute myeloid leukemia (AML), thus recapitulating the high risk of myeloid malignancies in FA patients better than Fancc-/-mice. We utilized hematopoietic stem cell transplantation to propagate Fancc-/-; Mad2+/- AML in irradiated healthy mice to model FANCC-deficient AMLs arising in the non-FA population. Compared to cells from Fancc-/- mice, those from Fancc-/-;Mad2+/- mice demonstrated an increase in mitotic errors but equivalent DNA cross-linker hypersensitivity, indicating that the cancer phenotype of Fancc-/-;Mad2+/- mice results from error-prone cell division and not exacerbation of the DNA damage repair defect. We found that FANCC enhances targeting of endogenous MAD2 to prometaphase kinetochores, suggesting a mechanism for how FANCC-dependent regulation of the spindle assembly checkpoint prevents chromosome mis-segregation. Whole-exome sequencing revealed similarities between human FA-associated myelodysplastic syndrome (MDS)/AML and the AML that developed in Fancc-/-; Mad2+/- mice. Together, these data illuminate the role of mitotic dysregulation in FA-pathway deficient malignancies in vivo, show how FANCC adjusts the spindle assembly checkpoint rheostat by regulating MAD2 kinetochore targeting in cell cycle-dependent manner, and establish two new mouse models for preclinical studies of AML.
Keywords: FANCC; Fanconi anemia; genomic instability; leukemia; spindle assembly checkpoint.
Copyright © 2021 Edwards, Mitchell, Abdul-Sater, Chan, Sun, Sheth, He, Jiang, Yuan, Sharma, Czader, Chin, Liu, de Cárcer, Nalepa, Broxmeyer, Clapp and Sierra Potchanant.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
A role for the Fanconi anemia C protein in maintaining the DNA damage-induced G2 checkpoint.J Biol Chem. 2004 Dec 3;279(49):50986-93. doi: 10.1074/jbc.M407160200. Epub 2004 Sep 17. J Biol Chem. 2004. PMID: 15377654
-
Leukemia and chromosomal instability in aged Fancc-/- mice.Exp Hematol. 2016 May;44(5):352-7. doi: 10.1016/j.exphem.2016.01.009. Epub 2016 Feb 6. Exp Hematol. 2016. PMID: 26860989 Free PMC article.
-
Mad2-independent spindle assembly checkpoint activation and controlled metaphase-anaphase transition in Drosophila S2 cells.Mol Biol Cell. 2007 Mar;18(3):850-63. doi: 10.1091/mbc.e06-07-0587. Epub 2006 Dec 20. Mol Biol Cell. 2007. PMID: 17182852 Free PMC article.
-
Complex Commingling: Nucleoporins and the Spindle Assembly Checkpoint.Cells. 2015 Nov 3;4(4):706-25. doi: 10.3390/cells4040706. Cells. 2015. PMID: 26540075 Free PMC article. Review.
-
Enhanced genomic instabilities caused by deregulated microtubule dynamics and chromosome segregation: a perspective from genetic studies in mice.Carcinogenesis. 2009 Sep;30(9):1469-74. doi: 10.1093/carcin/bgp081. Epub 2009 Apr 16. Carcinogenesis. 2009. PMID: 19372138 Free PMC article. Review.
Cited by
-
PLK1 Inhibition Induces Synthetic Lethality in Fanconi Anemia Pathway-Deficient Acute Myeloid Leukemia.Cancer Res Commun. 2025 Apr 1;5(4):648-667. doi: 10.1158/2767-9764.CRC-24-0260. Cancer Res Commun. 2025. PMID: 40111122 Free PMC article.
-
Mitotic DNA Synthesis in Untransformed Human Cells Preserves Common Fragile Site Stability via a FANCD2-Driven Mechanism That Requires HELQ.J Mol Biol. 2023 Nov 15;435(22):168294. doi: 10.1016/j.jmb.2023.168294. Epub 2023 Sep 28. J Mol Biol. 2023. PMID: 37777152 Free PMC article.
-
A New Multi-Color FISH Assay for Brush Biopsy-Based Detection of Chromosomal Aneuploidy in Oral (Pre)Cancer in Patients with Fanconi Anemia.Cancers (Basel). 2022 Jul 17;14(14):3468. doi: 10.3390/cancers14143468. Cancers (Basel). 2022. PMID: 35884529 Free PMC article.
-
Intervention of machine learning in bladder cancer research using multi-omics datasets: systematic review on biomarker identification.Discov Oncol. 2025 Jun 5;16(1):1010. doi: 10.1007/s12672-025-02734-6. Discov Oncol. 2025. PMID: 40471489 Free PMC article. Review.
-
Identification of a Hypomorphic FANCG Variant in Bernese Mountain Dogs.Genes (Basel). 2022 Sep 21;13(10):1693. doi: 10.3390/genes13101693. Genes (Basel). 2022. PMID: 36292578 Free PMC article.
References
-
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. . The Cbio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous