

Crosstalk Between ER Stress, Autophagy and Inflammation
- PMID: 34805224
- PMCID: PMC8602556
- DOI: 10.3389/fmed.2021.758311
Crosstalk Between ER Stress, Autophagy and Inflammation
Abstract
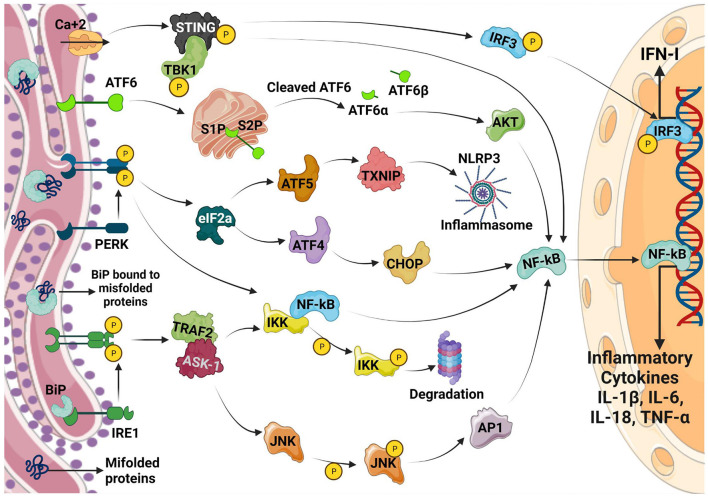
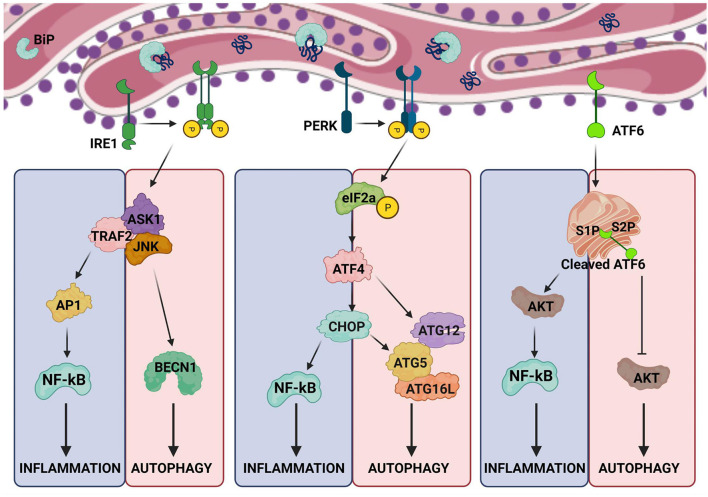
The endoplasmic reticulum (ER) is not only responsible for protein synthesis and folding but also plays a critical role in sensing cellular stress and maintaining cellular homeostasis. Upon sensing the accumulation of unfolded proteins due to perturbation in protein synthesis or folding, specific intracellular signaling pathways are activated, which are collectively termed as unfolded protein response (UPR). UPR expands the capacity of the protein folding machinery, decreases protein synthesis and enhances ER-associated protein degradation (ERAD) which degrades misfolded proteins through the proteasomes. More recent evidences suggest that UPR also amplifies cytokines-mediated inflammatory responses leading to pathogenesis of inflammatory diseases. UPR signaling also activates autophagy; a lysosome-dependent degradative pathwaythat has an extended capacity to degrade misfolded proteins and damaged ER. Thus, activation of autophagy limits inflammatory response and provides cyto-protection by attenuating ER-stress. Here we review the mechanisms that couple UPR, autophagy and cytokine-induced inflammation that can facilitate the development of novel therapeutic strategies to mitigate cellular stress and inflammation associated with various pathologies.
Keywords: ER-stress; autophagy; cytokines; inflammation; unfolded protein response.
Copyright © 2021 Chipurupalli, Samavedam and Robinson.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources