

Role of endothelial cells in pulmonary fibrosis via SREBP2 activation
- PMID: 34806652
- PMCID: PMC8663776
- DOI: 10.1172/jci.insight.125635
Role of endothelial cells in pulmonary fibrosis via SREBP2 activation
Abstract
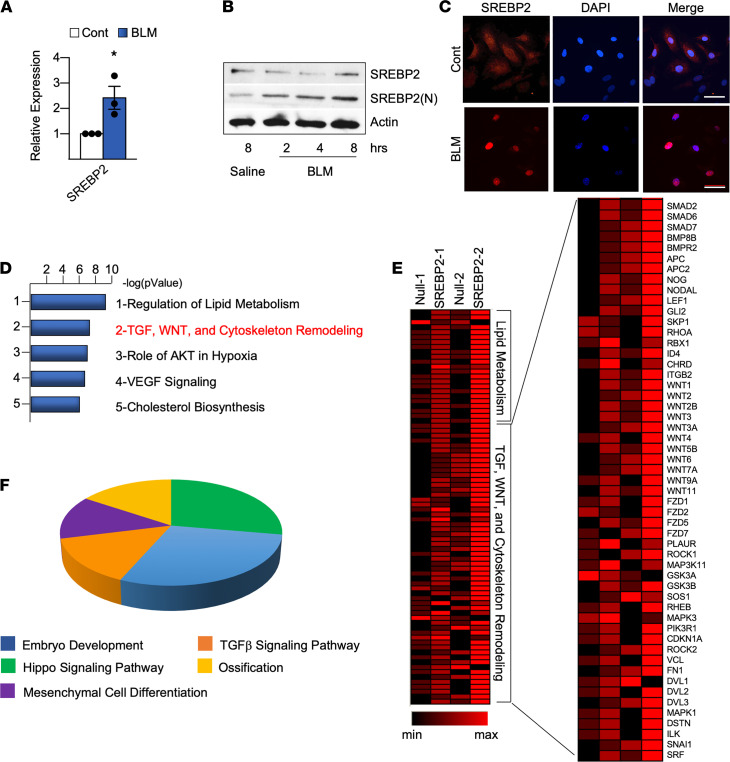
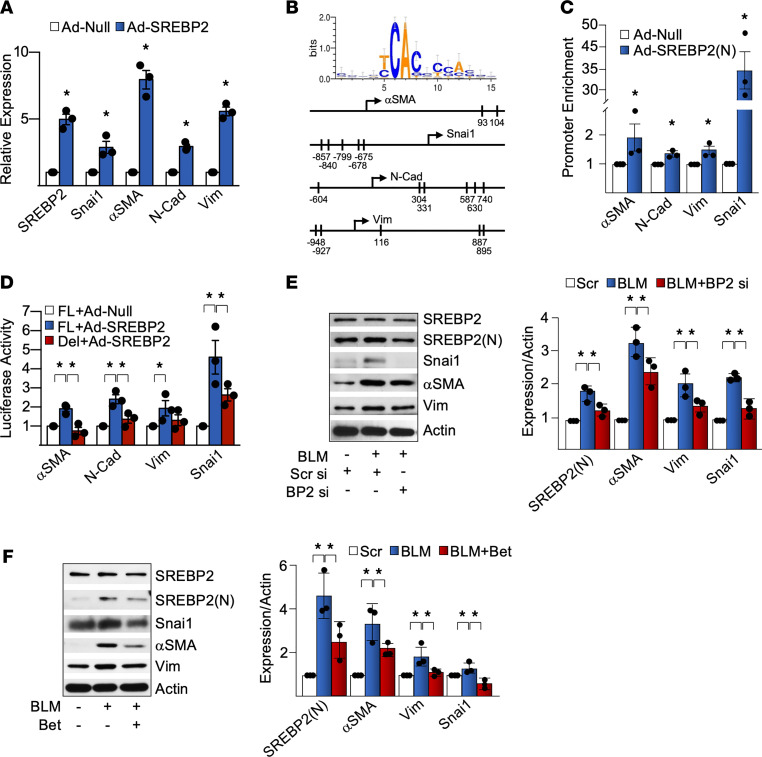
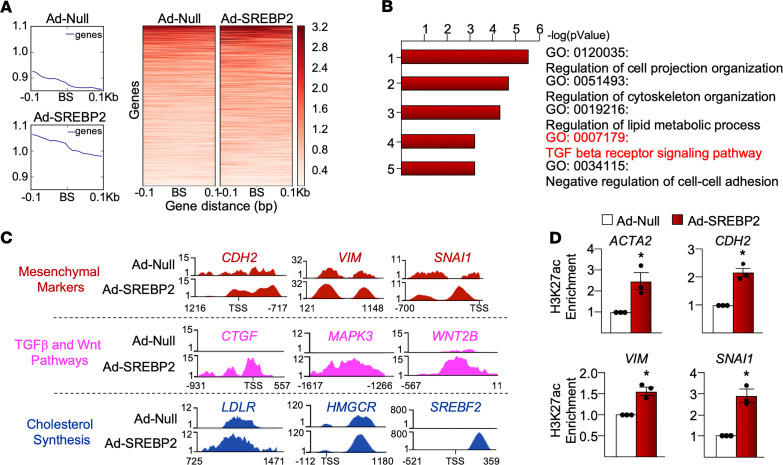
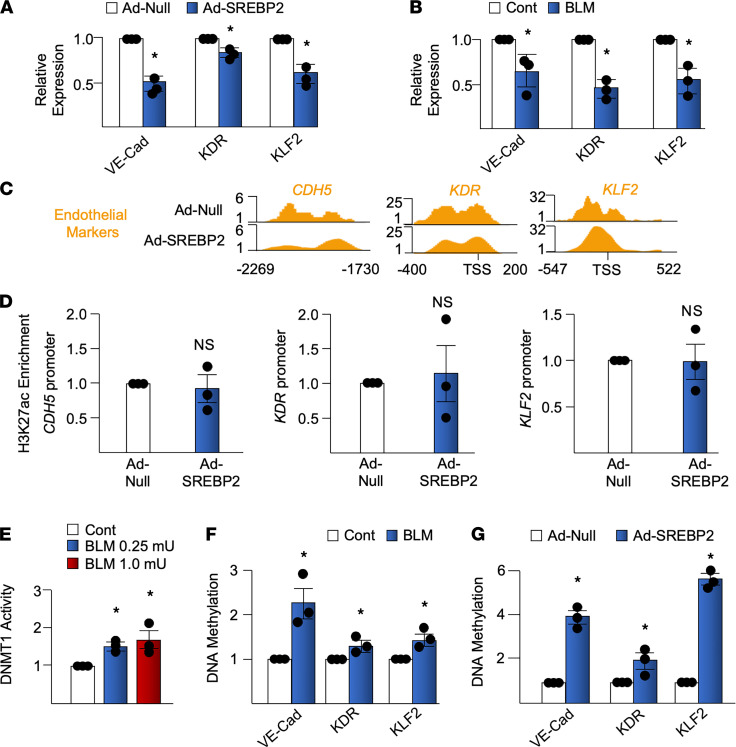
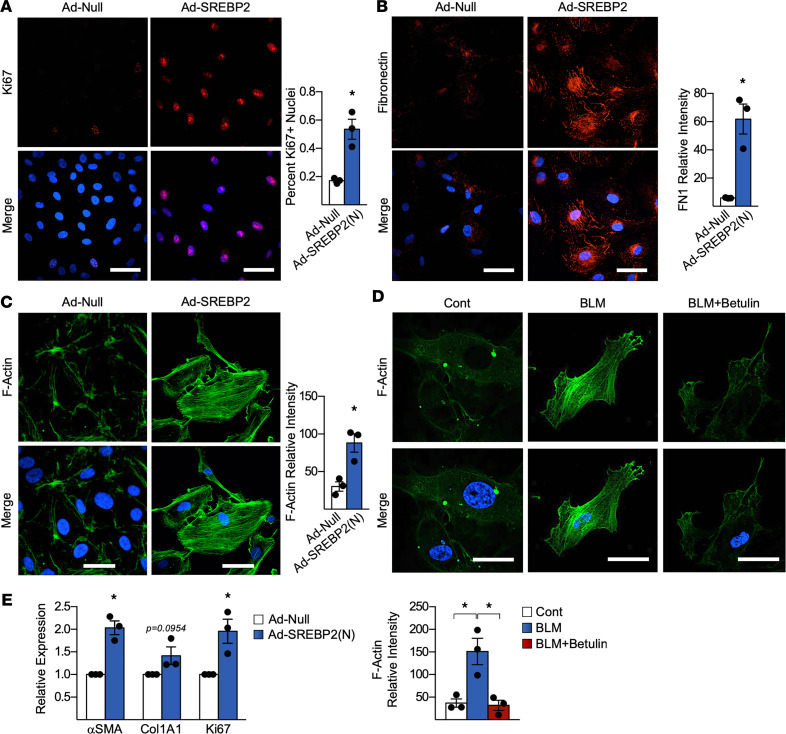
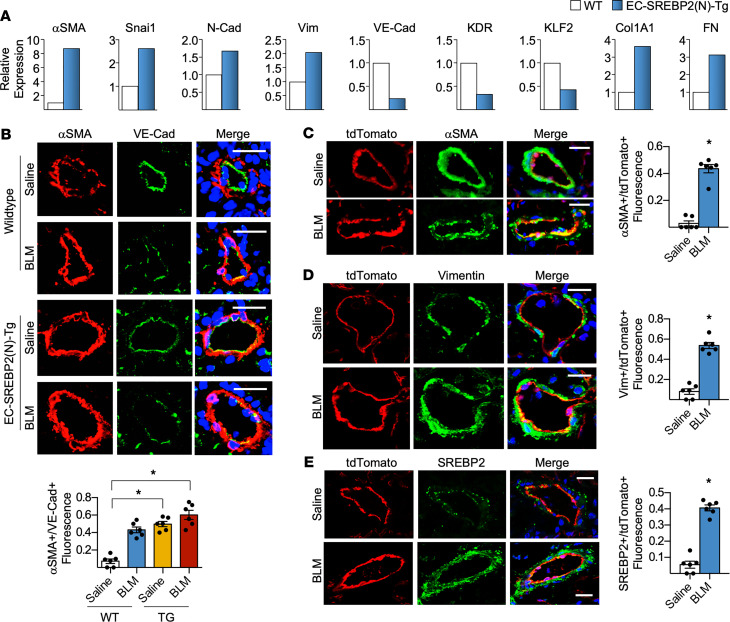
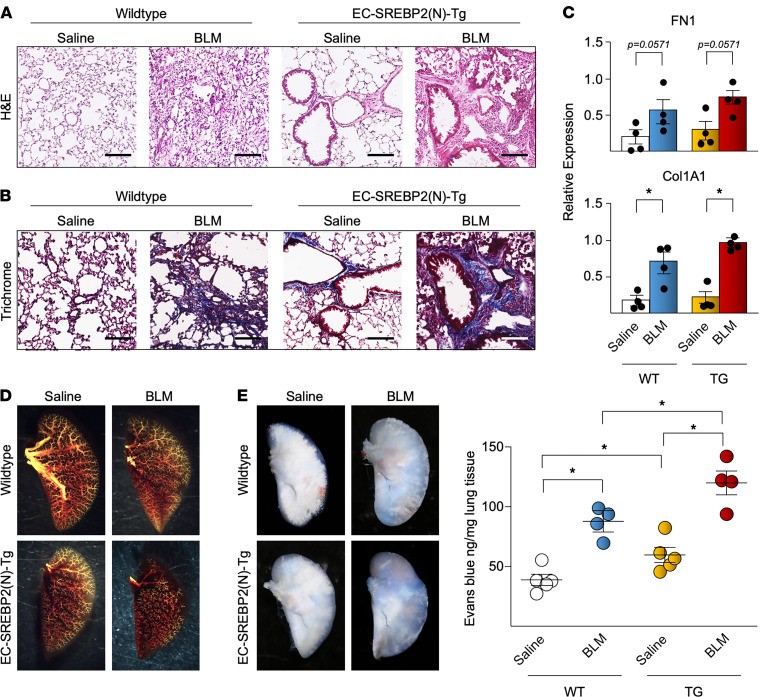
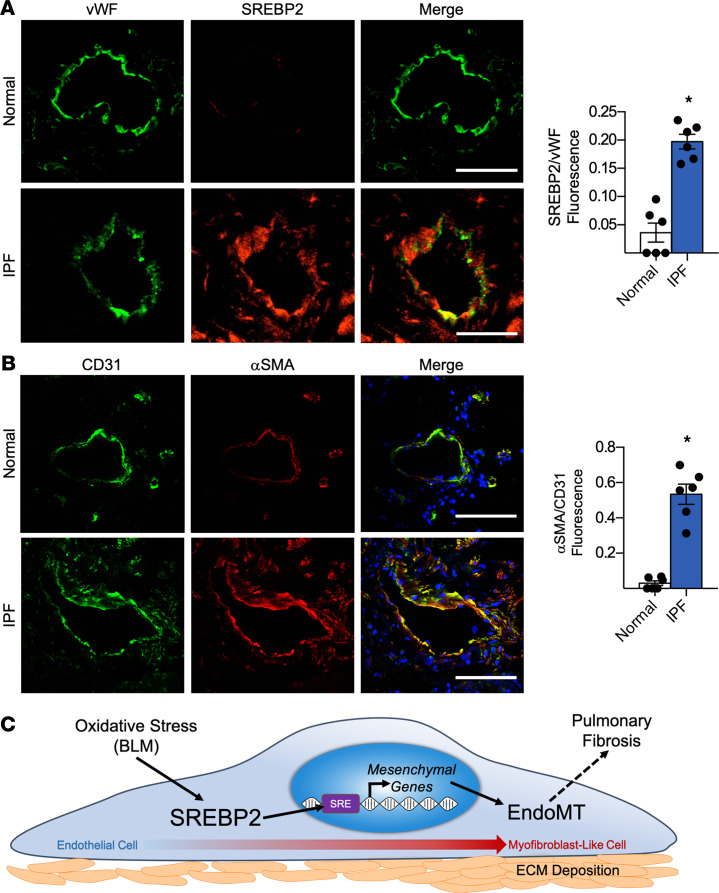
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease with limited treatment options. Despite endothelial cells (ECs) comprising 30% of the lung cellular composition, the role of EC dysfunction in pulmonary fibrosis (PF) remains unclear. We hypothesize that sterol regulatory element-binding protein 2 (SREBP2) plays a critical role in the pathogenesis of PF via EC phenotypic modifications. Transcriptome data demonstrate that SREBP2 overexpression in ECs led to the induction of the TGF, Wnt, and cytoskeleton remodeling gene ontology pathways and the increased expression of mesenchymal genes, such as snail family transcriptional repressor 1 (snai1), α-smooth muscle actin, vimentin, and neural cadherin. Furthermore, SREBP2 directly bound to the promoter regions and transactivated these mesenchymal genes. This transcriptomic change was associated with an epigenetic and phenotypic switch in ECs, leading to increased proliferation, stress fiber formation, and ECM deposition. Mice with endothelial-specific transgenic overexpression of SREBP2 (EC-SREBP2[N]-Tg mice) that were administered bleomycin to induce PF demonstrated exacerbated vascular remodeling and increased mesenchymal transition in the lung. SREBP2 was also found to be markedly increased in lung specimens from patients with IPF. These results suggest that SREBP2, induced by lung injury, can exacerbate PF in rodent models and in human patients with IPF.
Keywords: Endothelial cells; Fibrosis; Pulmonology; Vascular Biology.
Conflict of interest statement
Figures
Similar articles
-
Inflammatory stress signaling via NF-kB alters accessible cholesterol to upregulate SREBP2 transcriptional activity in endothelial cells.Elife. 2022 Aug 12;11:e79529. doi: 10.7554/eLife.79529. Elife. 2022. PMID: 35959888 Free PMC article.
-
Oxidative stress activates endothelial innate immunity via sterol regulatory element binding protein 2 (SREBP2) transactivation of microRNA-92a.Circulation. 2015 Mar 3;131(9):805-14. doi: 10.1161/CIRCULATIONAHA.114.013675. Epub 2014 Dec 30. Circulation. 2015. PMID: 25550450 Free PMC article.
-
SREBP2 regulates the endothelial response to cytokines via direct transcriptional activation of KLF6.J Lipid Res. 2023 Aug;64(8):100411. doi: 10.1016/j.jlr.2023.100411. Epub 2023 Jul 10. J Lipid Res. 2023. PMID: 37437844 Free PMC article.
-
Targeting endothelial cells: A novel strategy for pulmonary fibrosis treatment.Eur J Pharmacol. 2025 Jun 15;997:177472. doi: 10.1016/j.ejphar.2025.177472. Epub 2025 Mar 5. Eur J Pharmacol. 2025. PMID: 40054716 Review.
-
Injured Endothelial Cell: A Risk Factor for Pulmonary Fibrosis.Int J Mol Sci. 2023 May 14;24(10):8749. doi: 10.3390/ijms24108749. Int J Mol Sci. 2023. PMID: 37240093 Free PMC article. Review.
Cited by
-
The novel molecular mechanism of pulmonary fibrosis: insight into lipid metabolism from reanalysis of single-cell RNA-seq databases.Lipids Health Dis. 2024 Apr 3;23(1):98. doi: 10.1186/s12944-024-02062-8. Lipids Health Dis. 2024. PMID: 38570797 Free PMC article. Review.
-
Celastrol modulates IRS1 expression to alleviate ovarian aging and to enhance follicular development.Cell Biol Toxicol. 2025 Sep 2;41(1):129. doi: 10.1007/s10565-025-10079-7. Cell Biol Toxicol. 2025. PMID: 40897829 Free PMC article.
-
Biogenesis and Function of circRNAs in Pulmonary Fibrosis.Curr Gene Ther. 2024;24(5):395-409. doi: 10.2174/0115665232284076240207073542. Curr Gene Ther. 2024. PMID: 39005062 Review.
-
Ginsenoside Rg3 attenuates pulmonary fibrosis by inhibiting endothelial to mesenchymal transition.Anim Cells Syst (Seoul). 2023 Aug 4;27(1):159-170. doi: 10.1080/19768354.2023.2244549. eCollection 2023. Anim Cells Syst (Seoul). 2023. PMID: 37554358 Free PMC article.
-
Identification of the novel role of sterol regulatory element binding proteins (SREBPs) in mechanotransduction and intraocular pressure regulation.FASEB J. 2023 Nov;37(11):e23248. doi: 10.1096/fj.202301185R. FASEB J. 2023. PMID: 37823226 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
