

High-resolution epitope mapping and characterization of SARS-CoV-2 antibodies in large cohorts of subjects with COVID-19
- PMID: 34811480
- PMCID: PMC8608966
- DOI: 10.1038/s42003-021-02835-2
High-resolution epitope mapping and characterization of SARS-CoV-2 antibodies in large cohorts of subjects with COVID-19
Abstract
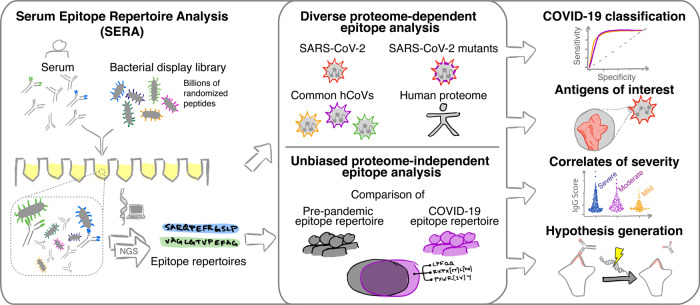
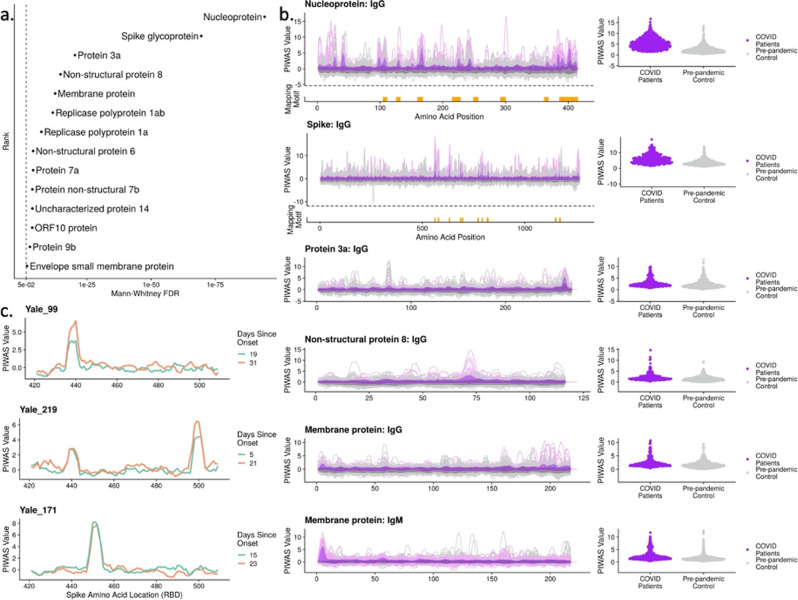
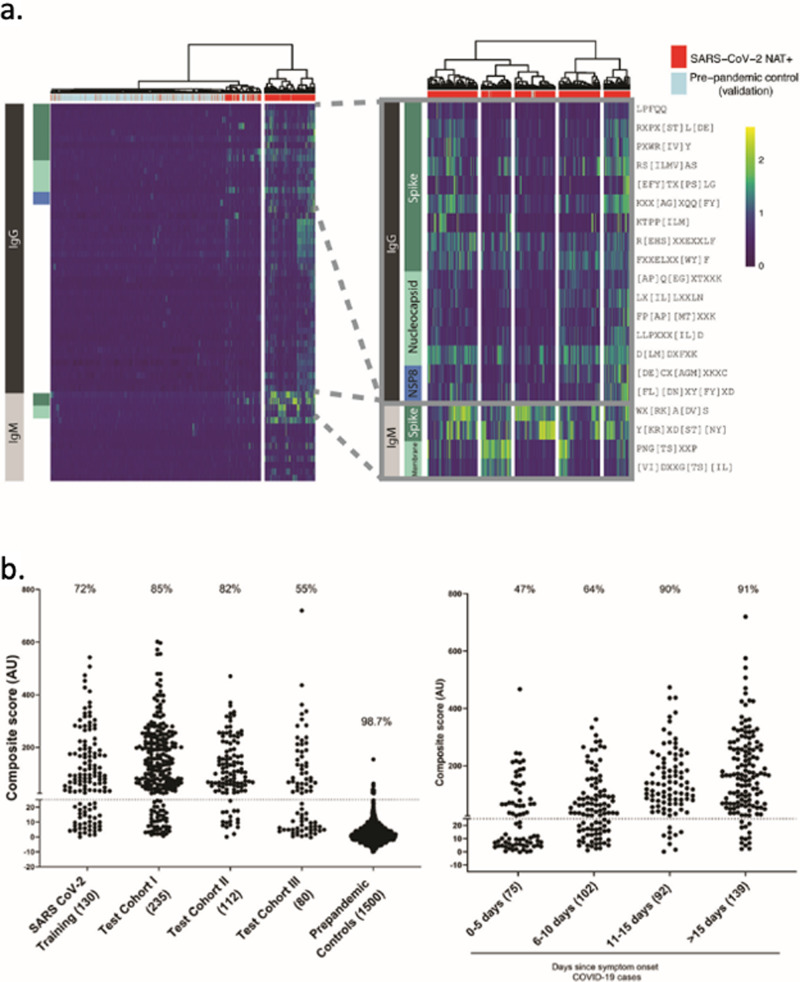
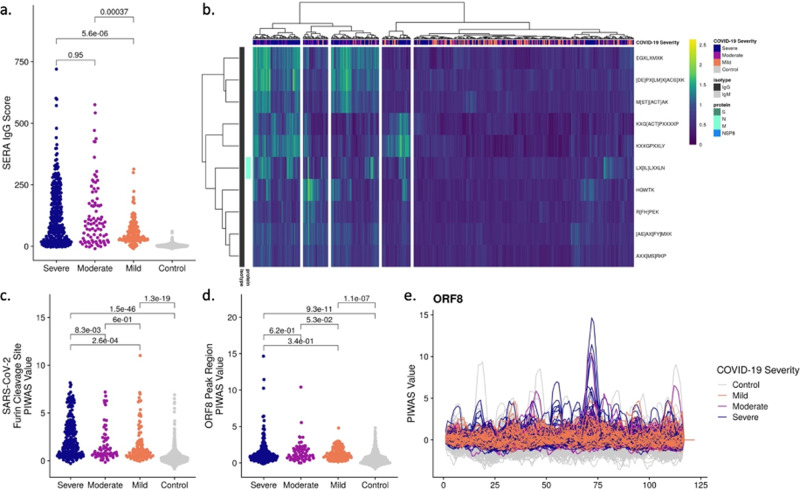
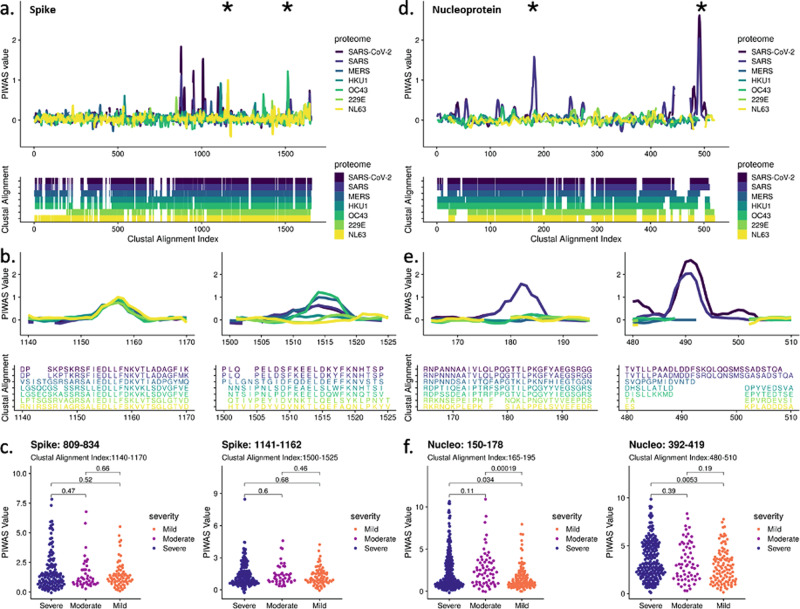
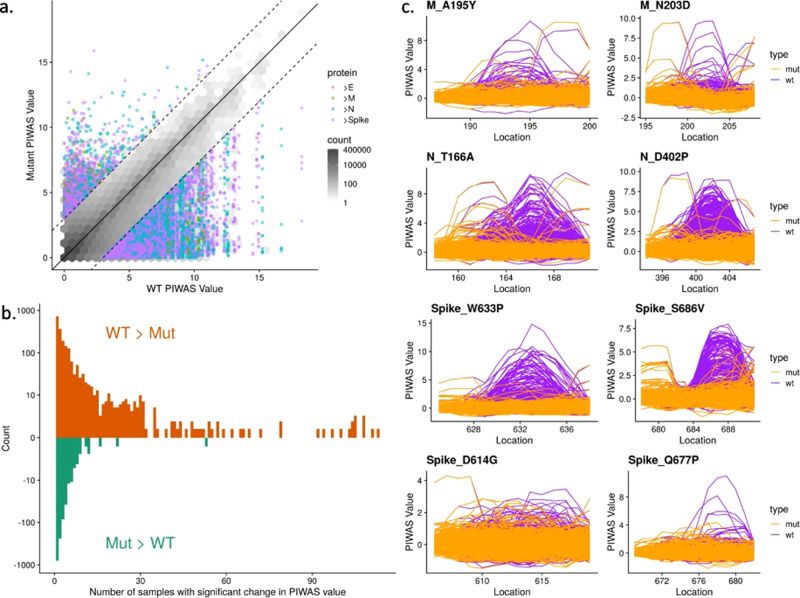
As Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) continues to spread, characterization of its antibody epitopes, emerging strains, related coronaviruses, and even the human proteome in naturally infected patients can guide the development of effective vaccines and therapies. Since traditional epitope identification tools are dependent upon pre-defined peptide sequences, they are not readily adaptable to diverse viral proteomes. The Serum Epitope Repertoire Analysis (SERA) platform leverages a high diversity random bacterial display library to identify proteome-independent epitope binding specificities which are then analyzed in the context of organisms of interest. When evaluating immune response in the context of SARS-CoV-2, we identify dominant epitope regions and motifs which demonstrate potential to classify mild from severe disease and relate to neutralization activity. We highlight SARS-CoV-2 epitopes that are cross-reactive with other coronaviruses and demonstrate decreased epitope signal for mutant SARS-CoV-2 strains. Collectively, the evolution of SARS-CoV-2 mutants towards reduced antibody response highlight the importance of data-driven development of the vaccines and therapies to treat COVID-19.
© 2021. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: employment and personal financial interests including stock options at Serimmune, In: W.A.H., K.K., J.B., E.B.J., P.S.D., A.D., M.J., G.J., B.M., J.R., J.R.S., R.W., M.Z., J.C.S. A.K. reports grant support from Regeneron and Merck for COVID-19 studies, contract and consulting fees from Tata for diagnostics development, honoraria for a COVID-19 lecture from Harvard University, and a research gift from Serimmune. Serimmune has also submitted the following US Patent Application 63/114,939 SARS-CoV-2 Serum Antibody Profiling SUI-009PR4. The remaining authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
