

Single-cell proteo-genomic reference maps of the hematopoietic system enable the purification and massive profiling of precisely defined cell states
- PMID: 34811546
- PMCID: PMC8642243
- DOI: 10.1038/s41590-021-01059-0
Single-cell proteo-genomic reference maps of the hematopoietic system enable the purification and massive profiling of precisely defined cell states
Abstract
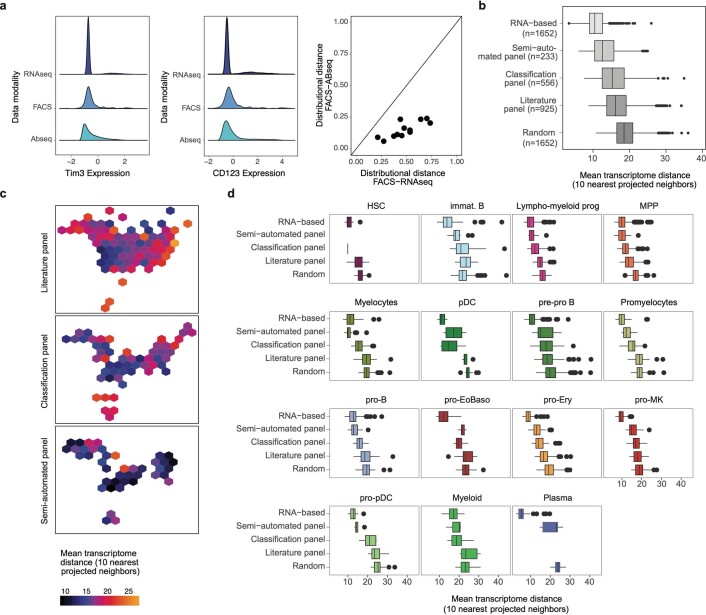
Single-cell genomics technology has transformed our understanding of complex cellular systems. However, excessive cost and a lack of strategies for the purification of newly identified cell types impede their functional characterization and large-scale profiling. Here, we have generated high-content single-cell proteo-genomic reference maps of human blood and bone marrow that quantitatively link the expression of up to 197 surface markers to cellular identities and biological processes across all main hematopoietic cell types in healthy aging and leukemia. These reference maps enable the automatic design of cost-effective high-throughput cytometry schemes that outperform state-of-the-art approaches, accurately reflect complex topologies of cellular systems and permit the purification of precisely defined cell states. The systematic integration of cytometry and proteo-genomic data enables the functional capacities of precisely mapped cell states to be measured at the single-cell level. Our study serves as an accessible resource and paves the way for a data-driven era in cytometry.
© 2021. The Author(s).
Conflict of interest statement
The oligo-coupled antibodies used in this study were a gift from BD Biosciences. The authors declare no other relevant conflicts of interest.
Figures








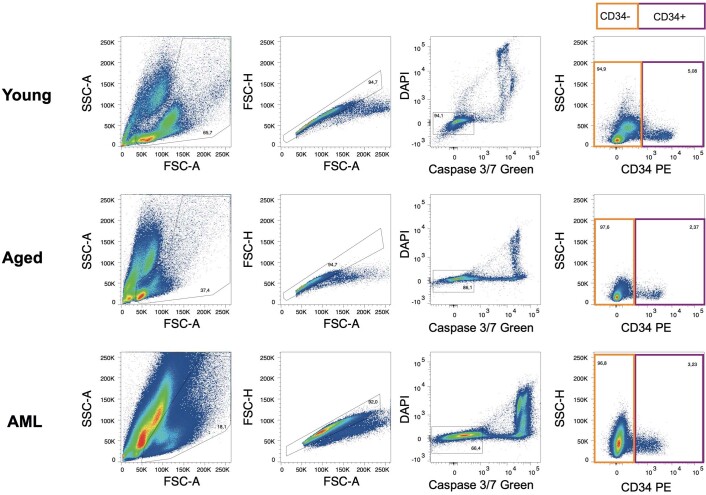
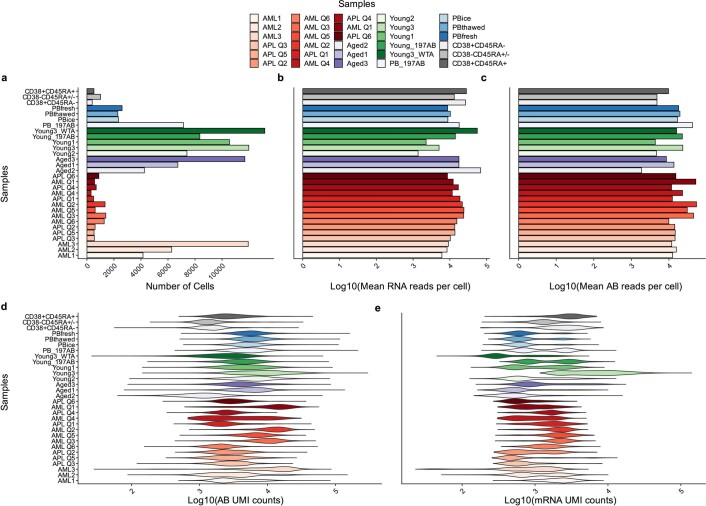


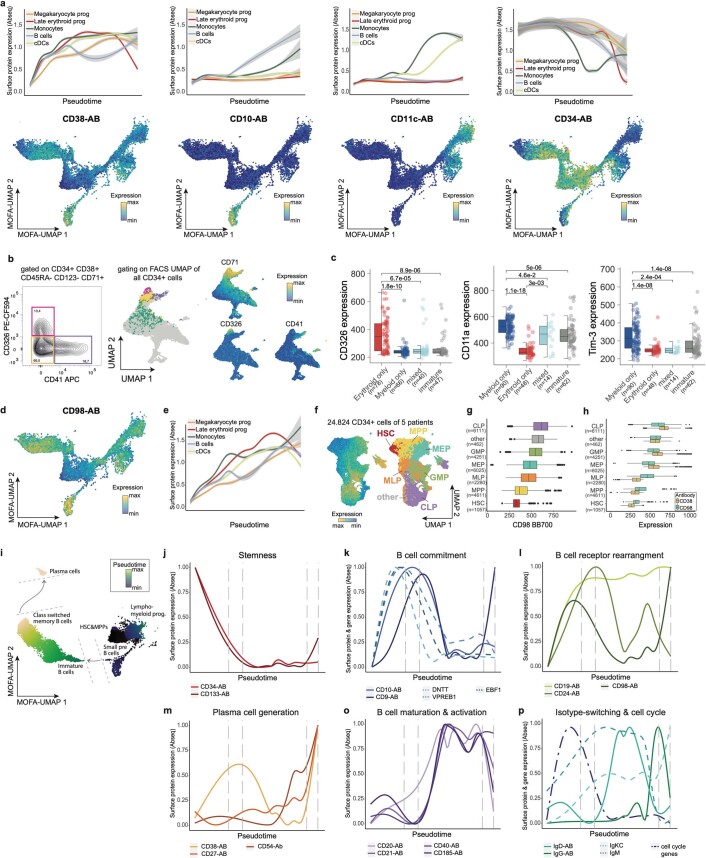

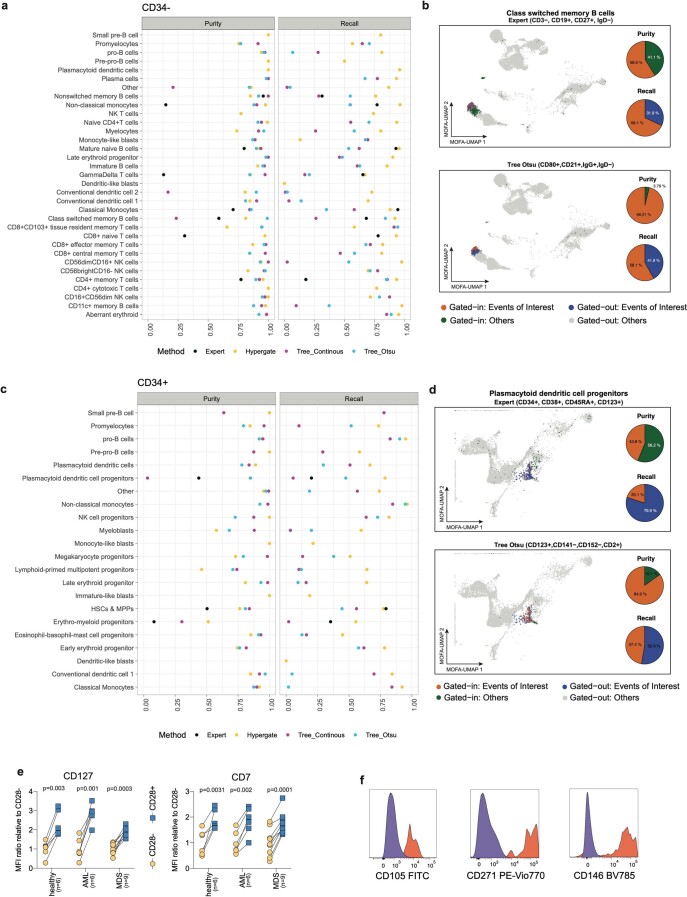

References
-
- Stuart T, Satija R. Integrative single-cell analysis. Nat. Rev. Genet. 2019;20:257–272. - PubMed
-
- Giladi A, Amit I. Single-cell genomics: a stepping stone for future immunology discoveries. Cell. 2018;172:14–21. - PubMed
-
- Han X, et al. Mapping the mouse cell atlas by Microwell-seq. Cell. 2018;172:1091–1107.e17. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
