

Enhancing CAR T function with the engineered secretion of C. perfringens neuraminidase
- PMID: 34813961
- PMCID: PMC8899523
- DOI: 10.1016/j.ymthe.2021.11.014
Enhancing CAR T function with the engineered secretion of C. perfringens neuraminidase
Abstract
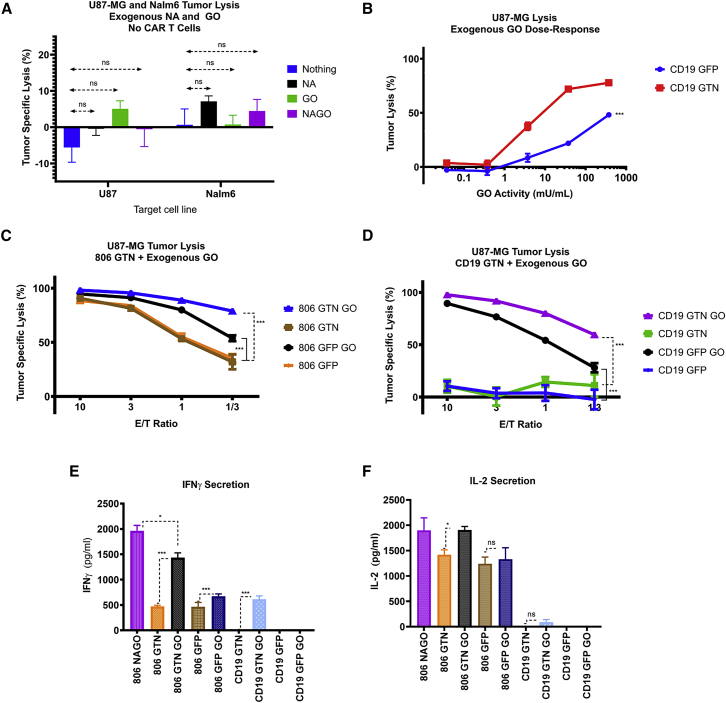
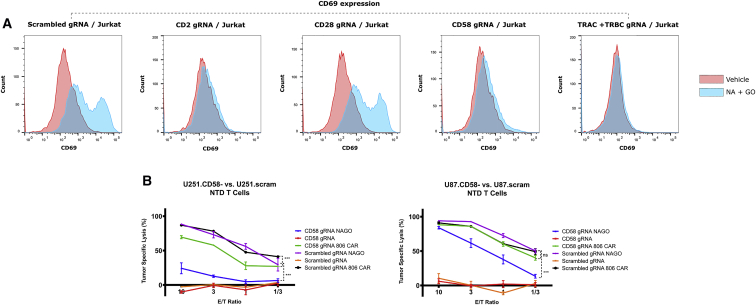
Prior to adoptive transfer, CAR T cells are activated, lentivirally infected with CAR transgenes, and expanded over 9 to 11 days. An unintended consequence of this process is the progressive differentiation of CAR T cells over time in culture. Differentiated T cells engraft poorly, which limits their ability to persist and provide sustained tumor control in hematologic as well as solid tumors. Solid tumors include other barriers to CAR T cell therapies, including immune and metabolic checkpoints that suppress effector function and durability. Sialic acids are ubiquitous surface molecules with known immune checkpoint functions. The enzyme C. perfringens neuraminidase (CpNA) removes sialic acid residues from target cells, with good activity at physiologic conditions. In combination with galactose oxidase (GO), NA has been found to stimulate T cell mitogenesis and cytotoxicity in vitro. Here we determine whether CpNA alone and in combination with GO promotes CAR T cell antitumor efficacy. We show that CpNA restrains CAR T cell differentiation during ex vivo culture, giving rise to progeny with enhanced therapeutic potential. CAR T cells expressing CpNA have superior effector function and cytotoxicity in vitro. In a Nalm-6 xenograft model of leukemia, CAR T cells expressing CpNA show enhanced antitumor efficacy. Arming CAR T cells with CpNA also enhanced tumor control in xenograft models of glioblastoma as well as a syngeneic model of melanoma. Given our findings, we hypothesize that charge repulsion via surface glycans is a regulatory parameter influencing differentiation. As T cells engage target cells within tumors and undergo constitutive activation through their CARs, critical thresholds of negative charge may impede cell-cell interactions underlying synapse formation and cytolysis. Removing the dense pool of negative cell-surface charge with CpNA is an effective approach to limit CAR T cell differentiation and enhance overall persistence and efficacy.
Keywords: CAR T cells; checkpoints; differentiation; glycoproteins; immunotherapy; neuraminidase; persistence.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.C.M. is an inventor on patent applications related to CAR technology and has received licensing royalties from Novartis corporation; S.G. and M.C.M. are inventors on patent applications related to methods of manufacturing CAR T cells. D.M.O., Z.B., L.J., R.T., and V.B. are inventors on patents related to CAR T cells that have been filed by the University of Pennsylvania. The other authors declare no financial or other conflicts of interest.
Figures




References
-
- Majzner R.G., Mackall C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019;25:1341–1355. - PubMed
-
- Dusoswa S.A., Verhoeff J., Abels E., Méndez-Huergo S.P., Croci D.O., Kuijper L.H., de Miguel E., Wouters V.M.C.J., Best M.G., Rodriguez E., et al. Glioblastomas exploit truncated O-linked glycans for local and distant immune modulation via the macrophage galactose-type lectin. Proc. Natl. Acad. Sci. U S A. 2020;117:3693–3703. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
