

Spliceator: multi-species splice site prediction using convolutional neural networks
- PMID: 34814826
- PMCID: PMC8609763
- DOI: 10.1186/s12859-021-04471-3
Spliceator: multi-species splice site prediction using convolutional neural networks
Abstract
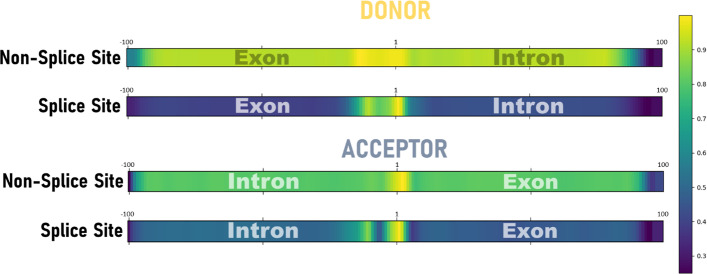
Background: Ab initio prediction of splice sites is an essential step in eukaryotic genome annotation. Recent predictors have exploited Deep Learning algorithms and reliable gene structures from model organisms. However, Deep Learning methods for non-model organisms are lacking.
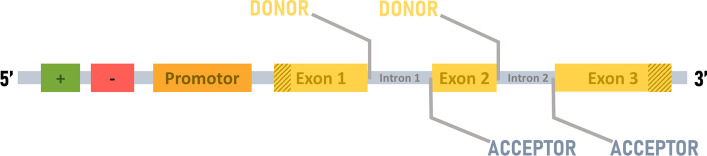
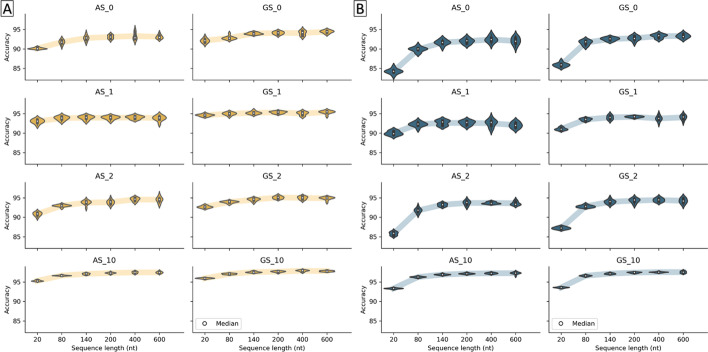
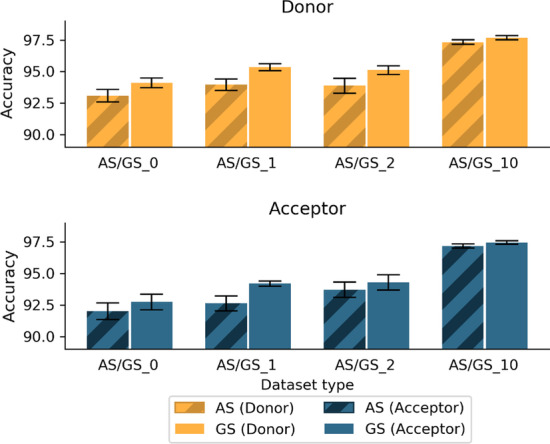
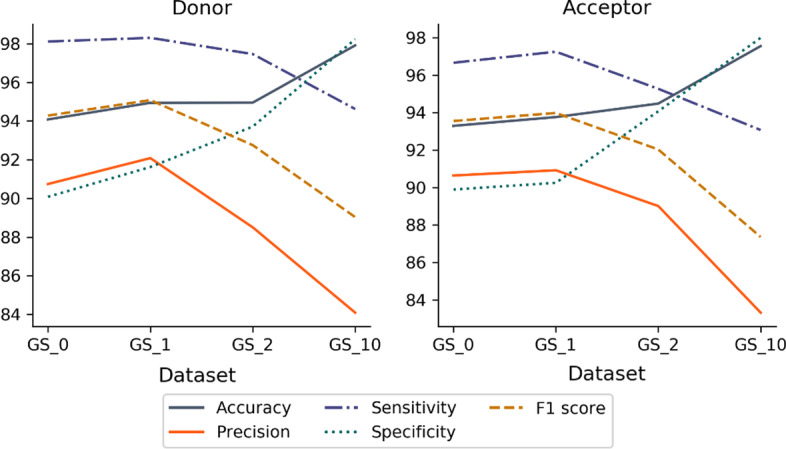
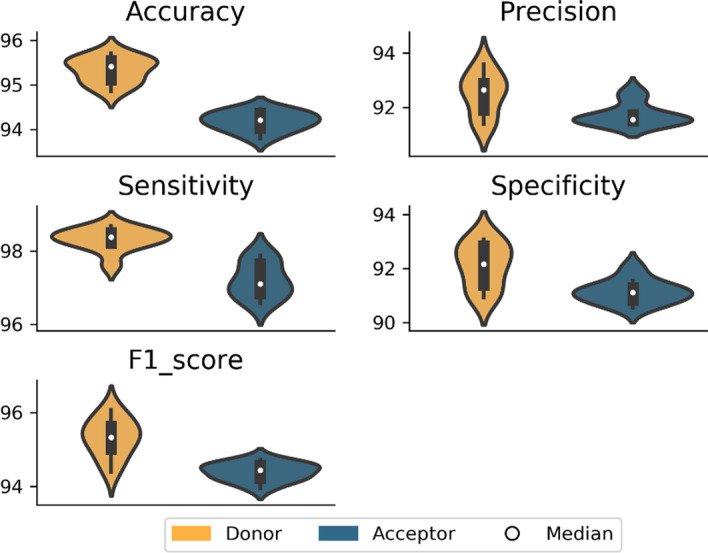
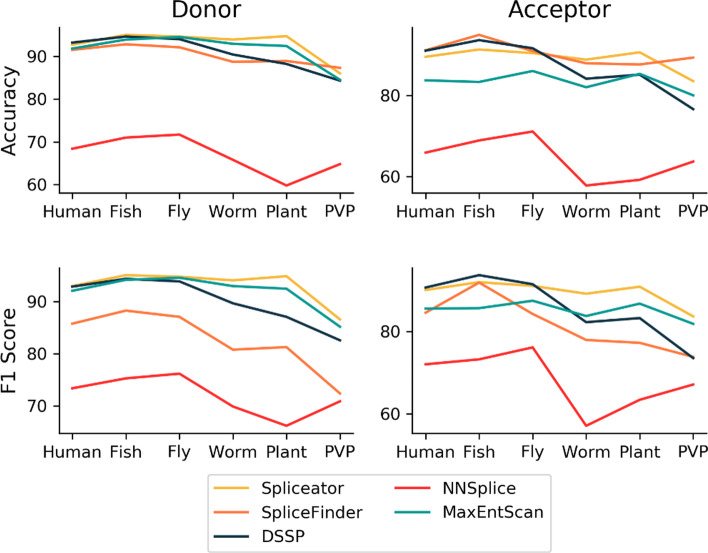
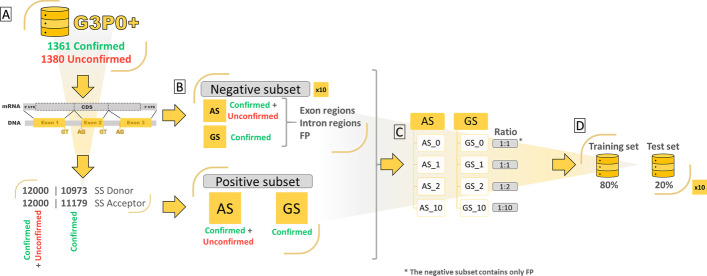
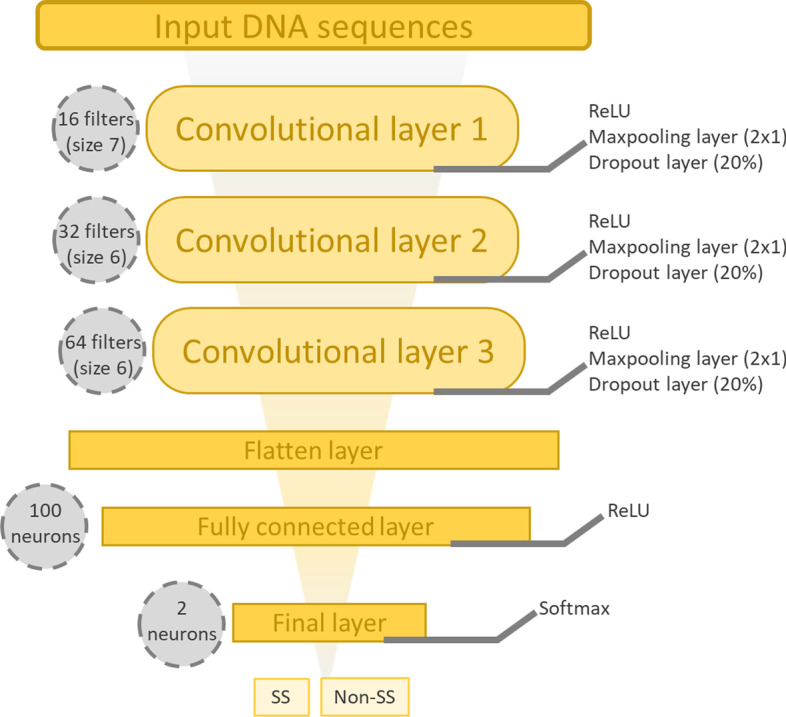
Results: We developed Spliceator to predict splice sites in a wide range of species, including model and non-model organisms. Spliceator uses a convolutional neural network and is trained on carefully validated data from over 100 organisms. We show that Spliceator achieves consistently high accuracy (89-92%) compared to existing methods on independent benchmarks from human, fish, fly, worm, plant and protist organisms.
Conclusions: Spliceator is a new Deep Learning method trained on high-quality data, which can be used to predict splice sites in diverse organisms, ranging from human to protists, with consistently high accuracy.
Keywords: Convolutional neural network; Data quality; Deep learning; Genome annotation; Splice site prediction.
© 2021. The Author(s).
Conflict of interest statement
JDT is a member of the editorial board (Associate Editor) of this journal. The authors declare they have no other competing interests.
Figures
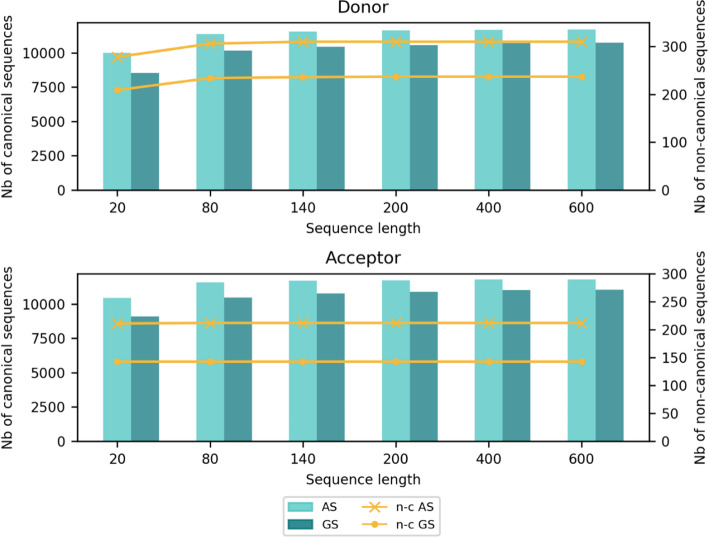
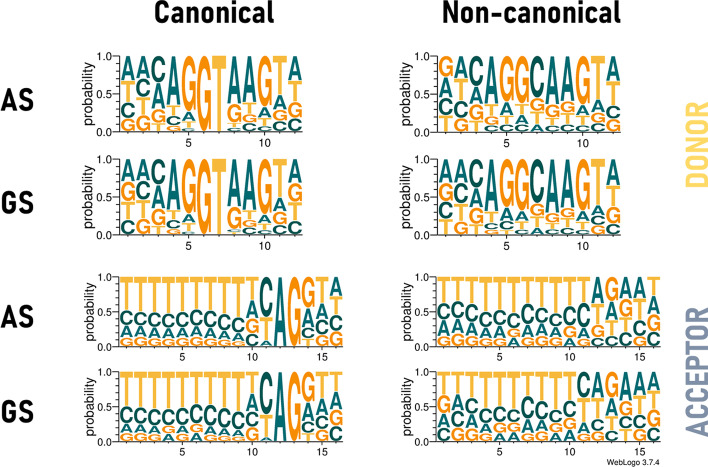
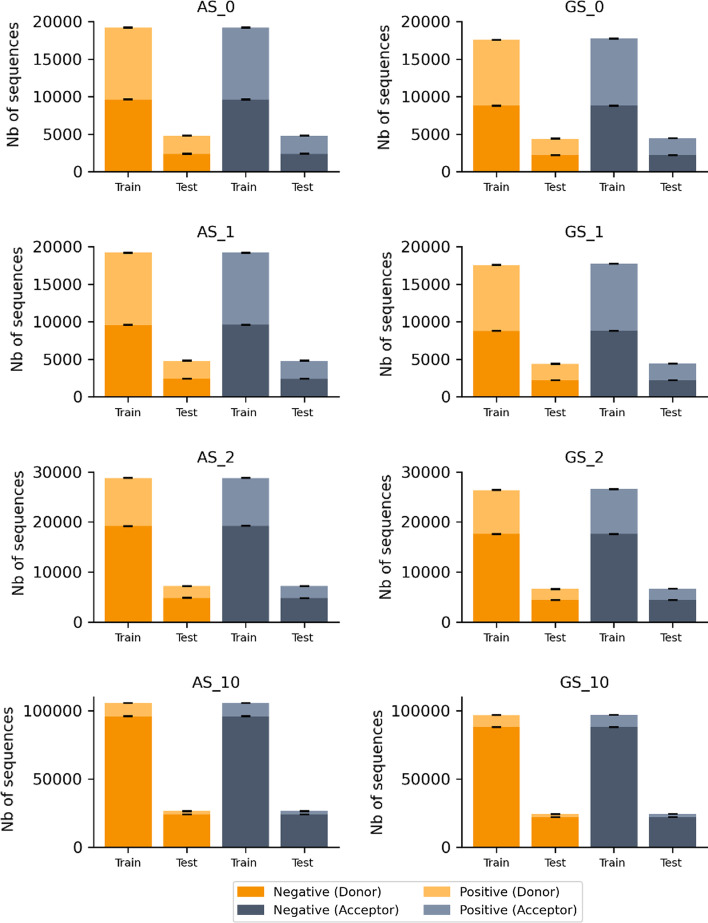
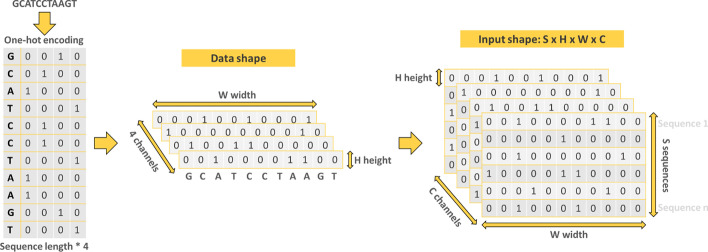
References
-
- Thibaud-Nissen F, DiCuccio M, Hlavina W, Kimchi A, Kitts PA, Murphy TD, et al. P8008 The NCBI eukaryotic genome annotation pipeline. J Anim Sci. 2016;94:184–184.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
