

Individualized VDJ recombination predisposes the available Ig sequence space
- PMID: 34815307
- PMCID: PMC8647828
- DOI: 10.1101/gr.275373.121
Individualized VDJ recombination predisposes the available Ig sequence space
Abstract
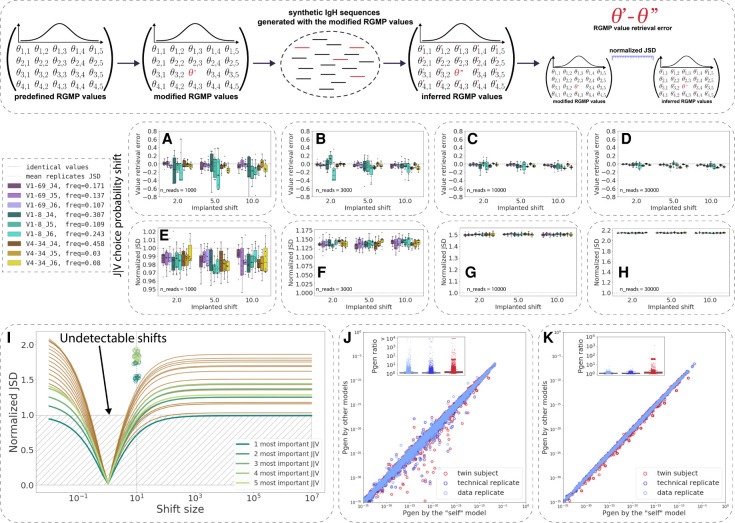
The process of recombination between variable (V), diversity (D), and joining (J) immunoglobulin (Ig) gene segments determines an individual's naive Ig repertoire and, consequently, (auto)antigen recognition. VDJ recombination follows probabilistic rules that can be modeled statistically. So far, it remains unknown whether VDJ recombination rules differ between individuals. If these rules differed, identical (auto)antigen-specific Ig sequences would be generated with individual-specific probabilities, signifying that the available Ig sequence space is individual specific. We devised a sensitivity-tested distance measure that enables inter-individual comparison of VDJ recombination models. We discovered, accounting for several sources of noise as well as allelic variation in Ig sequencing data, that not only unrelated individuals but also human monozygotic twins and even inbred mice possess statistically distinguishable immunoglobulin recombination models. This suggests that, in addition to genetic, there is also nongenetic modulation of VDJ recombination. We demonstrate that population-wide individualized VDJ recombination can result in orders of magnitude of difference in the probability to generate (auto)antigen-specific Ig sequences. Our findings have implications for immune receptor-based individualized medicine approaches relevant to vaccination, infection, and autoimmunity.
© 2021 Slabodkin et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
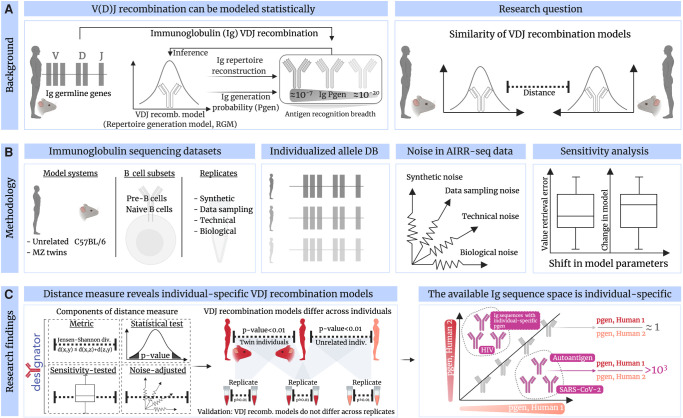
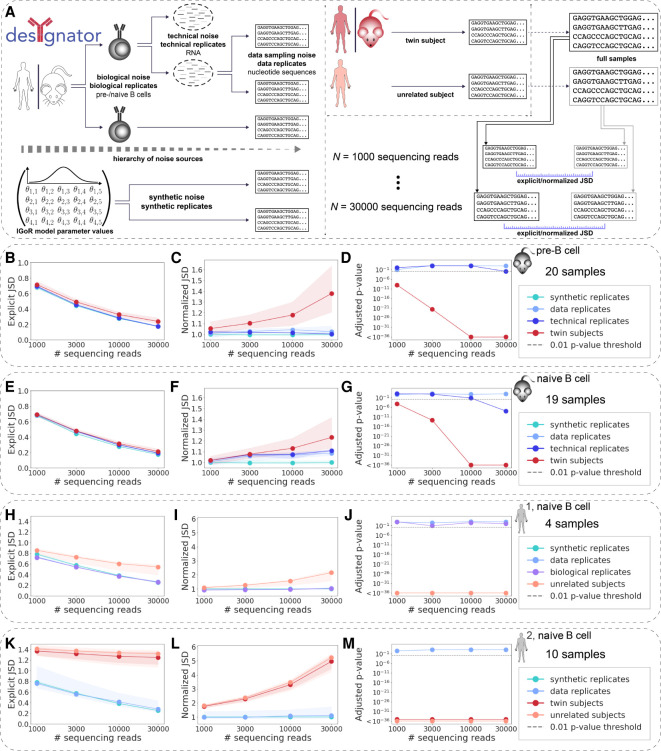
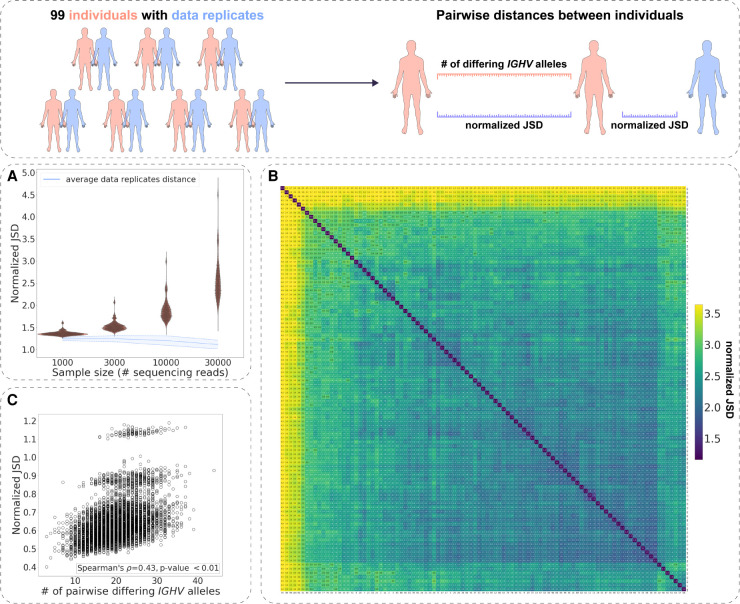
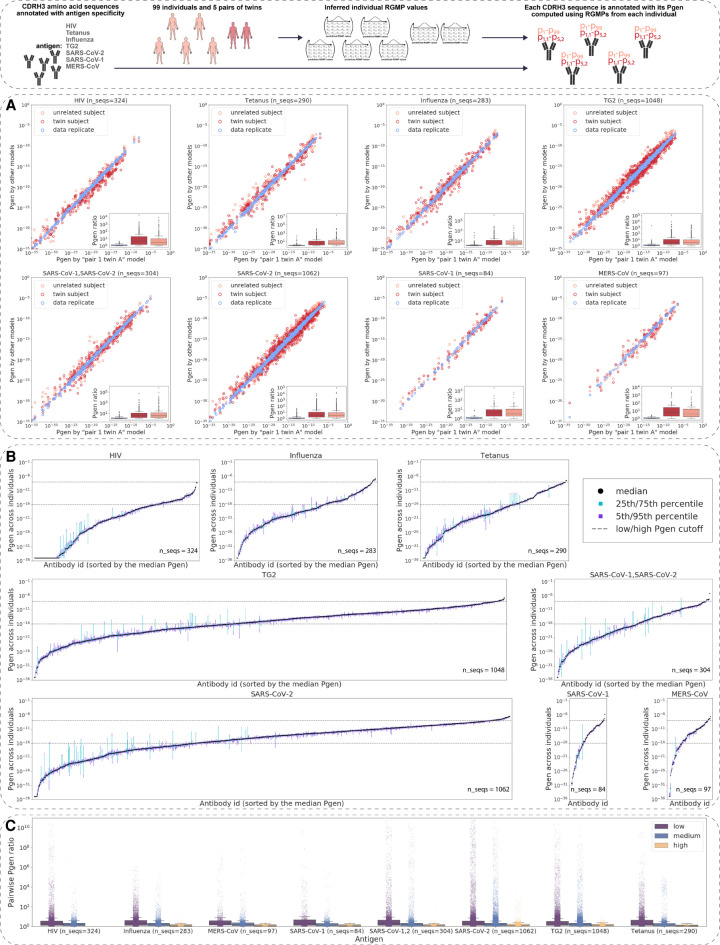
References
-
- Arora R, Burke HM, Arnaout R. 2018. Immunological diversity with similarity. bioRxiv 10.1101/483131 - DOI
-
- Avnir Y, Watson CT, Glanville J, Peterson EC, Tallarico AS, Bennett AS, Qin K, Fu Y, Huang C-Y, Beigel JH, et al. 2016. IGHV1-69 polymorphism modulates anti-influenza antibody repertoires, correlates with IGHV utilization shifts and varies by ethnicity. Sci Rep 6: 20842. 10.1038/srep20842 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources