

Parkinson's Disease Modification Through Abl Kinase Inhibition: An Opportunity
- PMID: 34816484
- PMCID: PMC8770606
- DOI: 10.1002/mds.28858
Parkinson's Disease Modification Through Abl Kinase Inhibition: An Opportunity
Abstract
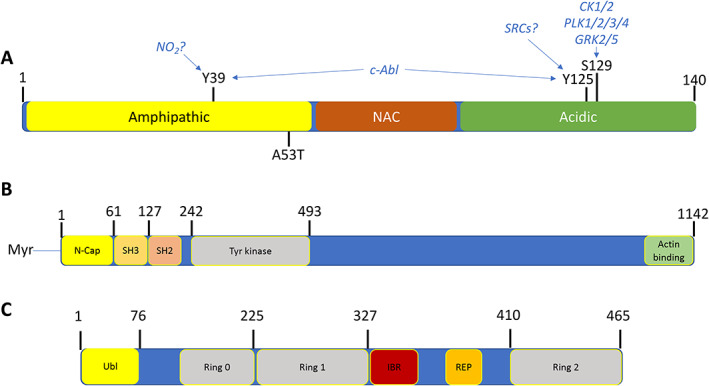
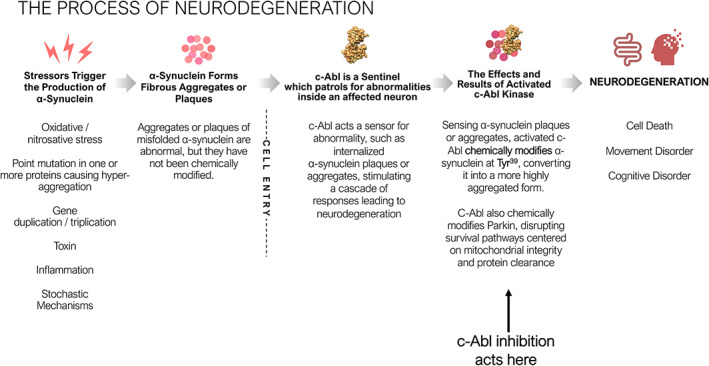
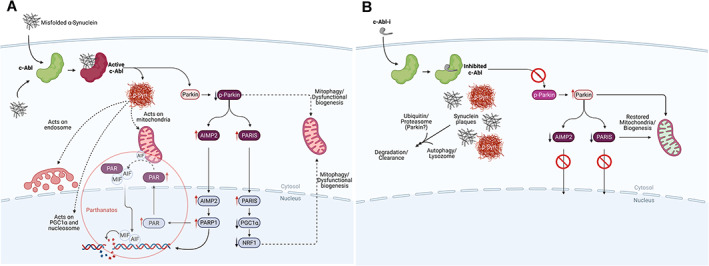
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease of the central nervous system, with an estimated 5 000 000 cases worldwide. Historically characterized by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta, PD pathology is now known to be widespread and to affect serotonin, cholinergic and norepinephrine neurons as well as nerve cells in the olfactory system, cerebral hemisphere, brain stem, spinal cord, and peripheral autonomic nervous system. PD pathology is characterized by the accumulation of misfolded α-synuclein, which is thought to play a critical role in the etiopathogenesis of the disease. Animal models of PD suggest that activation of the Abelson tyrosine kinase (c-Abl) plays an essential role in the initiation and progression of α-synuclein pathology and neurodegeneration. These studies demonstrate that internalization of misfolded α-synuclein activates c-Abl, which phosphorylates α-synuclein and promotes α-synuclein pathology within the affected neurons. Additionally, c-Abl inactivates parkin, disrupting mitochondrial quality control and biogenesis, promoting neurodegeneration. Post-mortem studies of PD patients demonstrate increased levels of tyrosine phosphorylated α-synuclein, consistent with the activation of c-Abl in human disease. Although the c-Abl inhibitor nilotinib failed to demonstrate clinical benefit in two double-blind trials, novel c-Abl inhibitors have been developed that accumulate in the brain and may inhibit c-Abl at saturating levels. These novel inhibitors have demonstrated benefits in animal models of PD and have now entered clinical development. Here, we review the role of c-Abl in the neurodegenerative disease process and consider the translational potential of c-Abl inhibitors from model studies to disease-modifying therapies for Parkinson's disease. © 2021 Inhibikase Therapeutics, Inc. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson Movement Disorder Society.
Keywords: Abelson tyrosine kinase; Parkinson's disease; disease-modification.
© 2021 Inhibikase Therapeutics, Inc. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson Movement Disorder Society.
Figures
References
-
- Bach JP, Ziegler U, Deuschl G, et al. Projected numbers of people with movement disorders in the years 2030 and 2050. Mov Disord 2011;26:2286–2290. - PubMed
-
- Olanow CW, Stern MB, Sethi K. Scientific and clinical basis for the treatment of PD – 2009. Neurology 2009;72(21 suppl 4):S1–S136. - PubMed
-
- Jellinger KA. Neuropathology of sporadic Parkinson's disease: evaluation and changes of concepts. Mov Disord 2012;27:8–30. - PubMed
-
- Schapira AHV, Chaudhuri KR, Jenner P. Non‐motor features of Parkinson disease. Nat Rev Neurosci 2017;18:435–450. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
