

Reconstitution of human atlastin fusion activity reveals autoinhibition by the C terminus
- PMID: 34817557
- PMCID: PMC8624677
- DOI: 10.1083/jcb.202107070
Reconstitution of human atlastin fusion activity reveals autoinhibition by the C terminus
Abstract
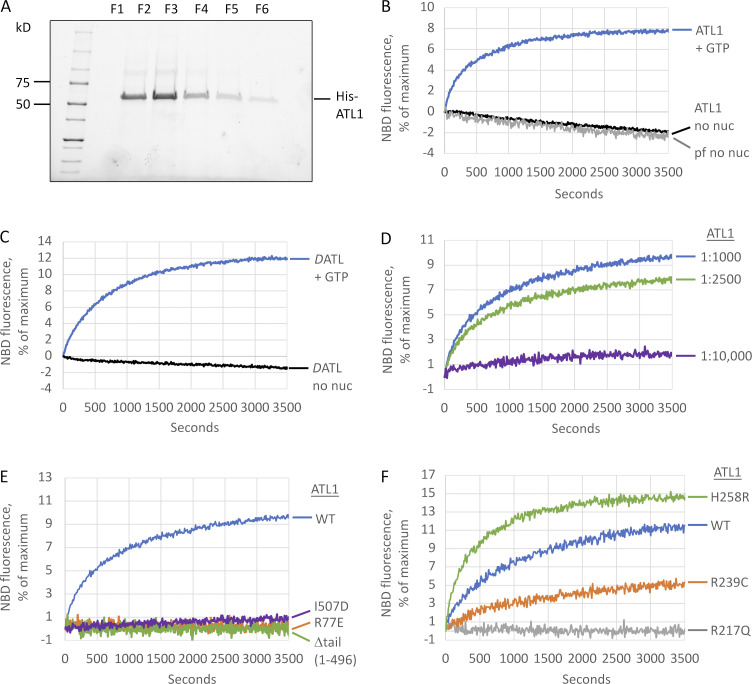
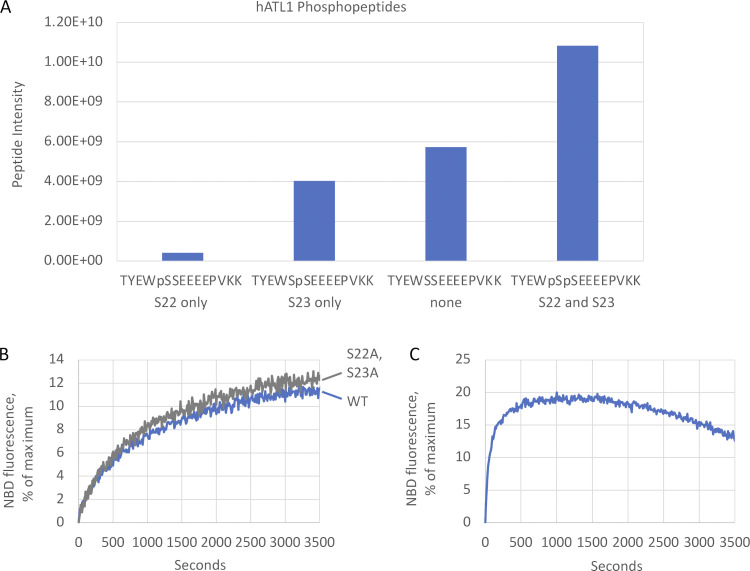
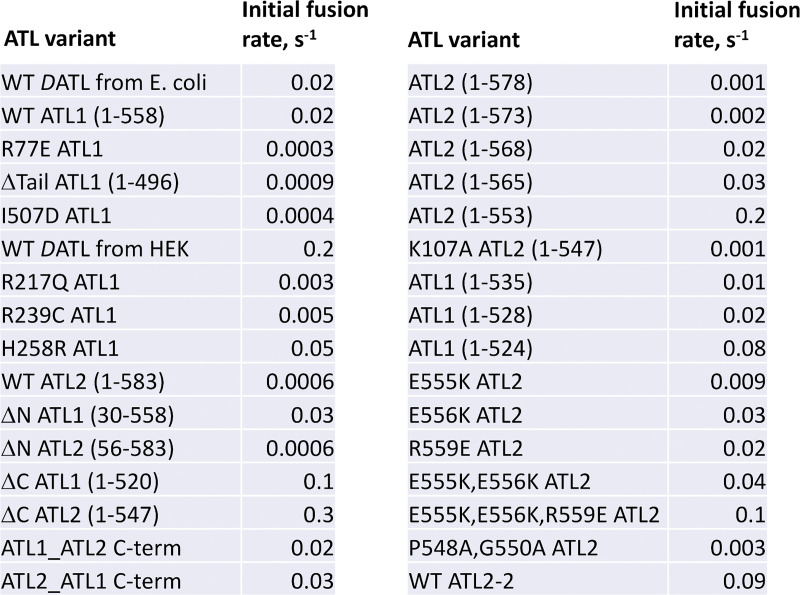
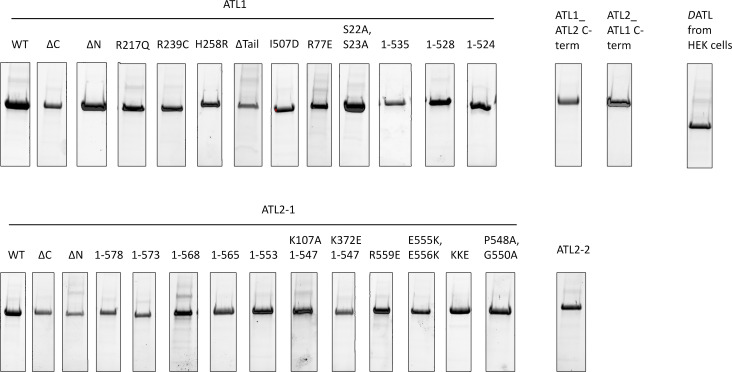
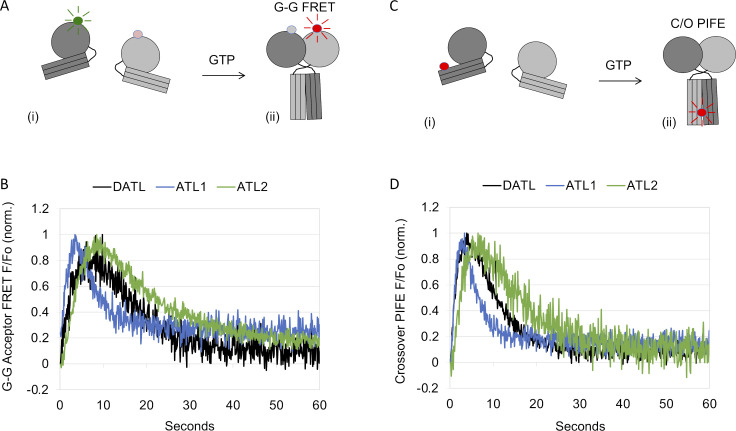
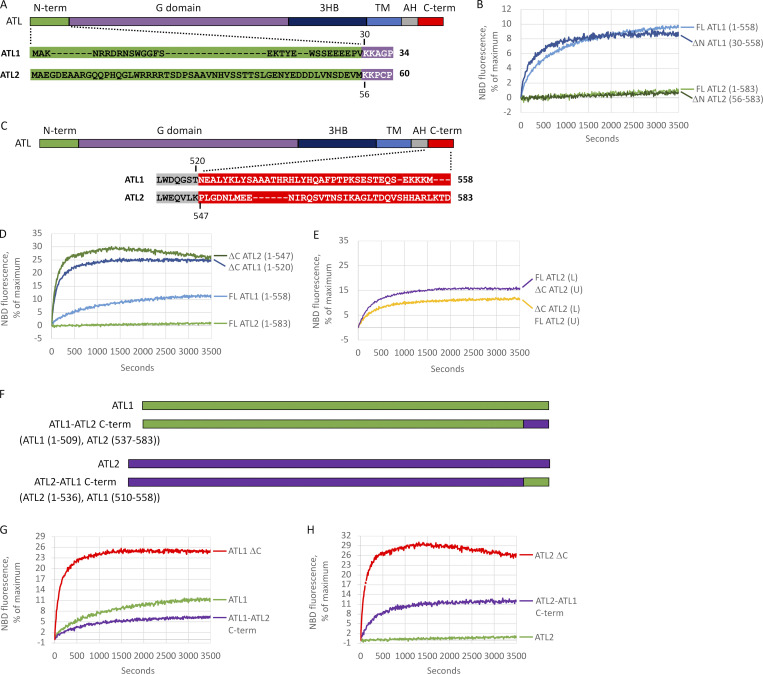
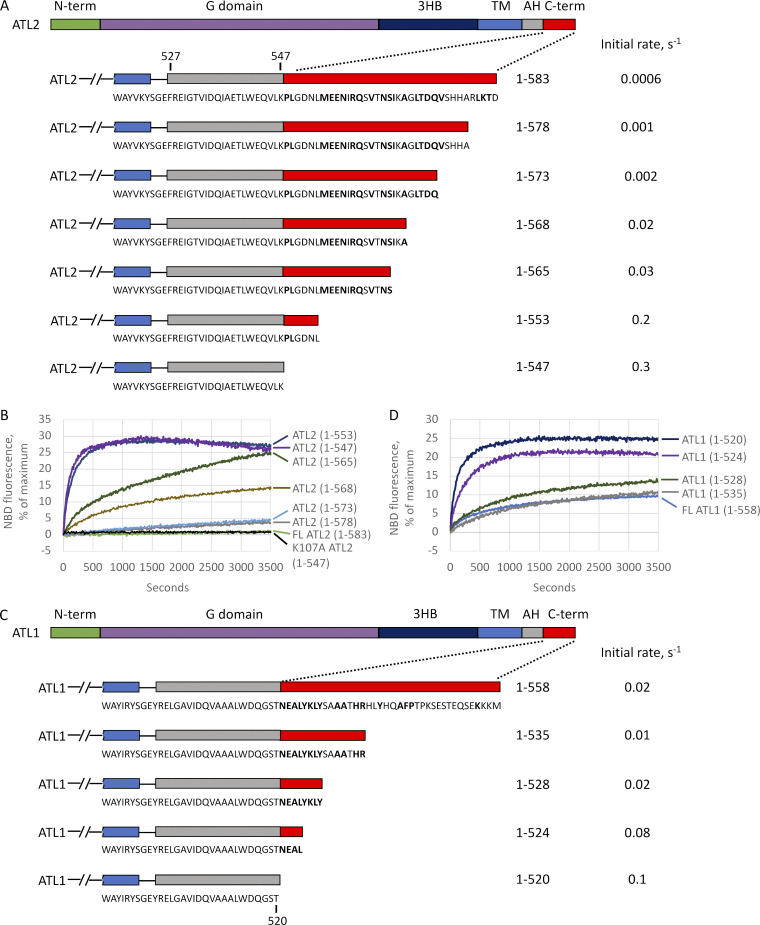
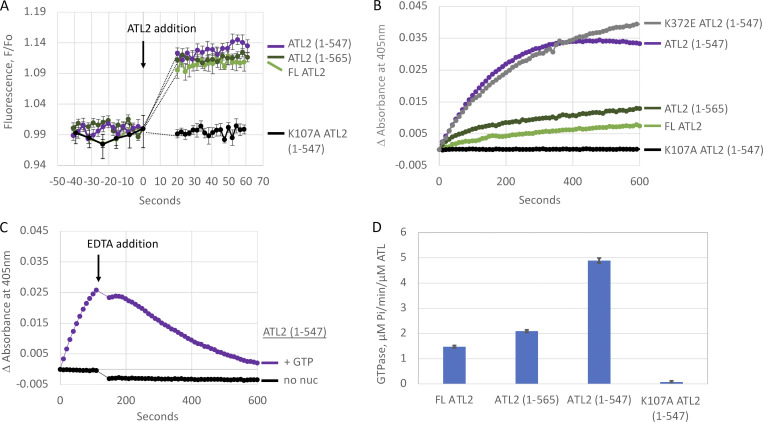
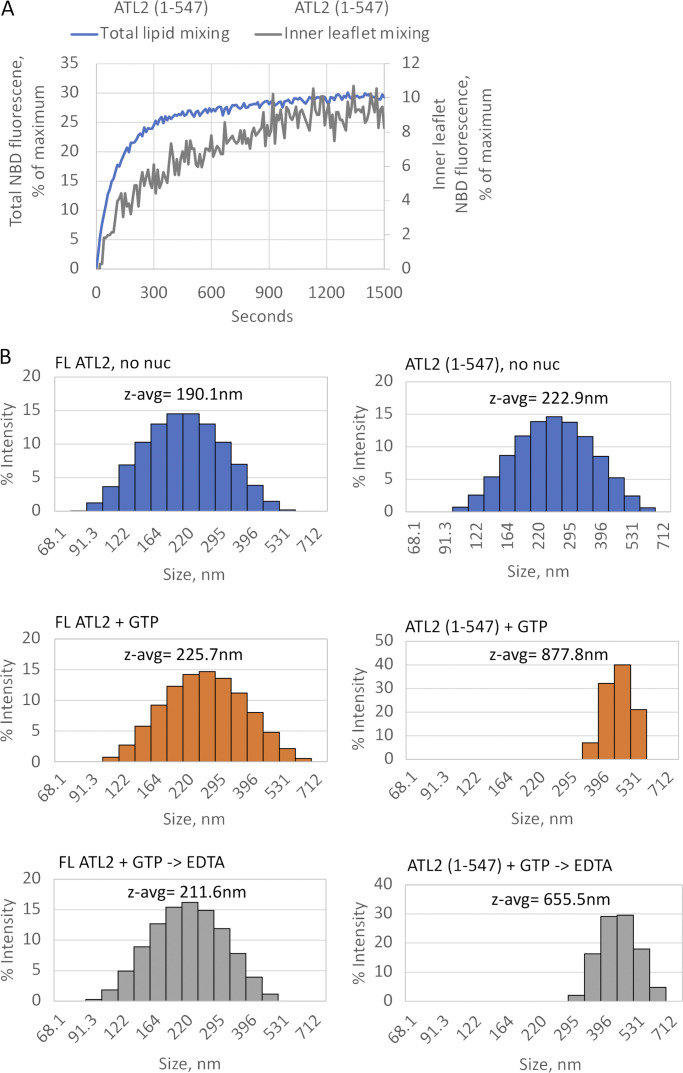
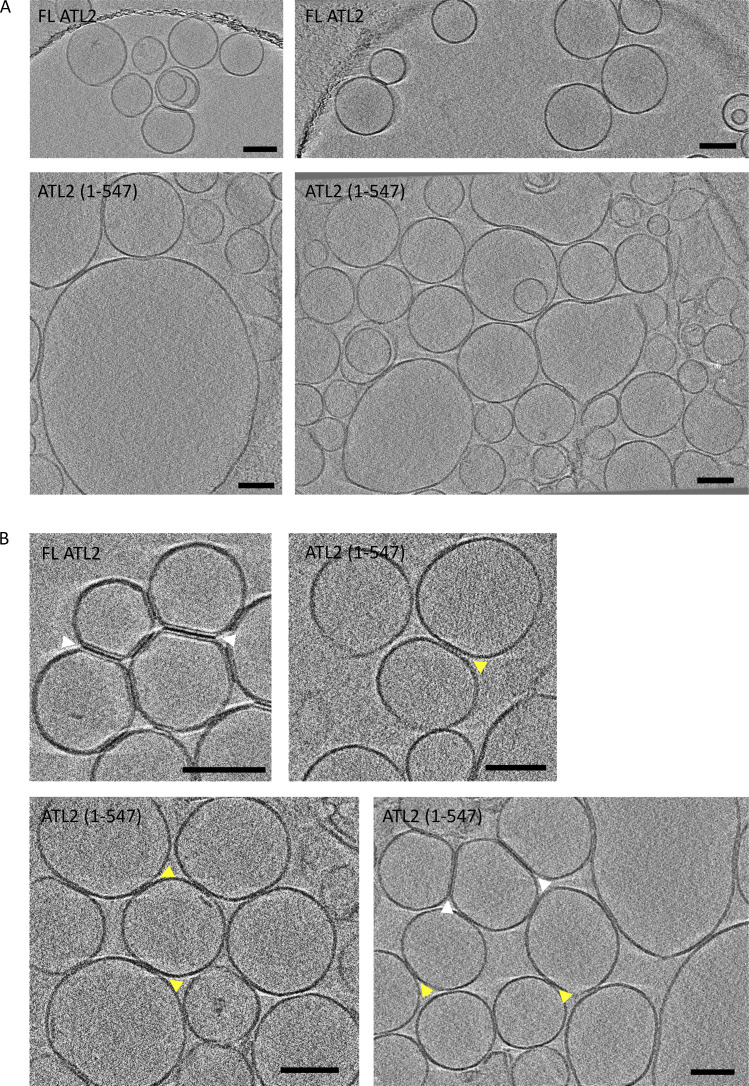
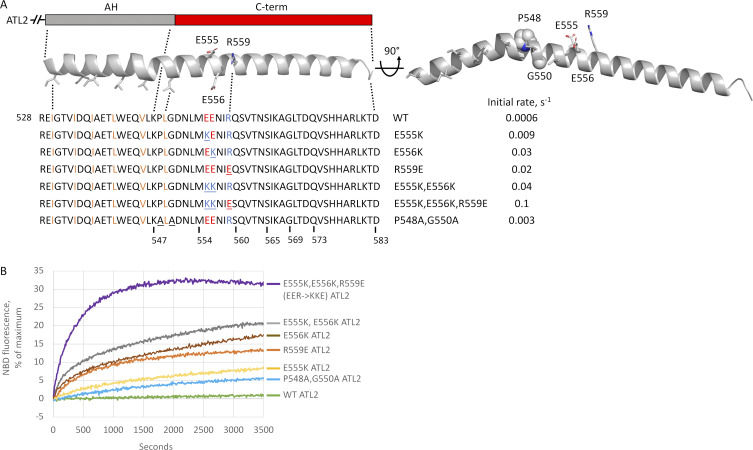
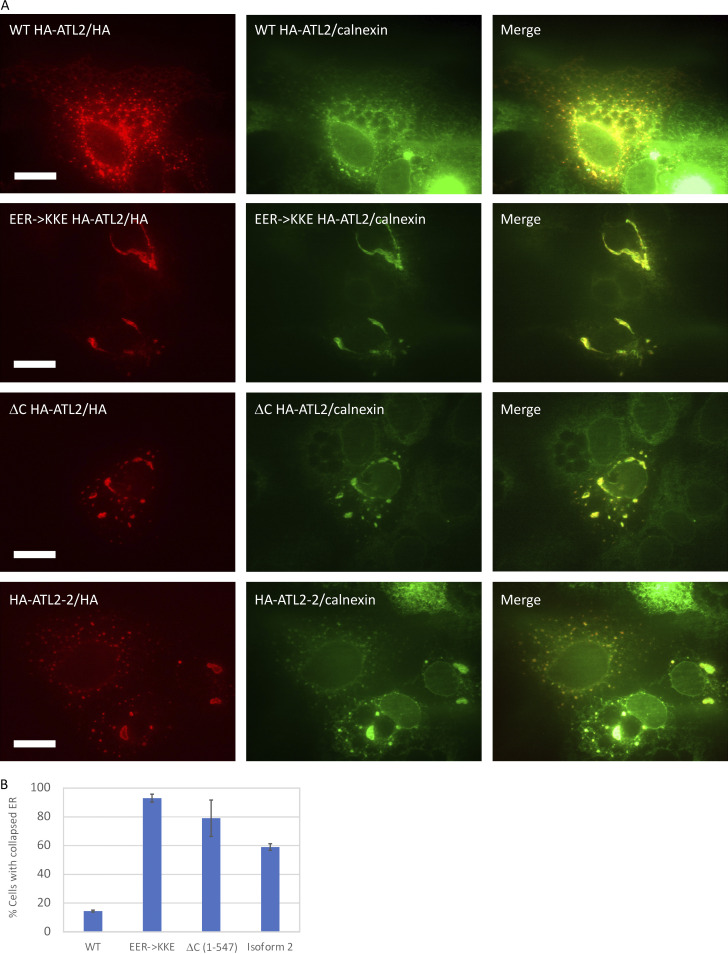
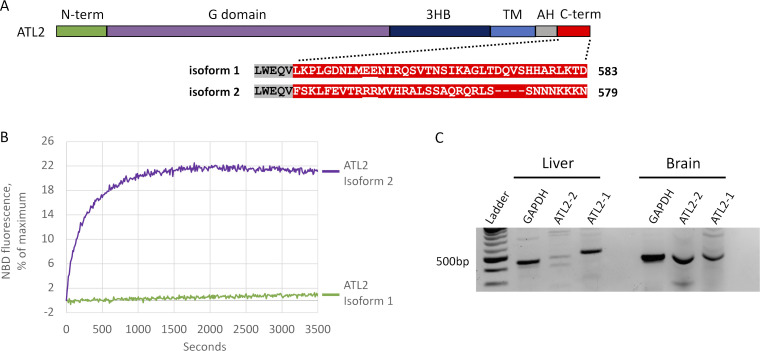
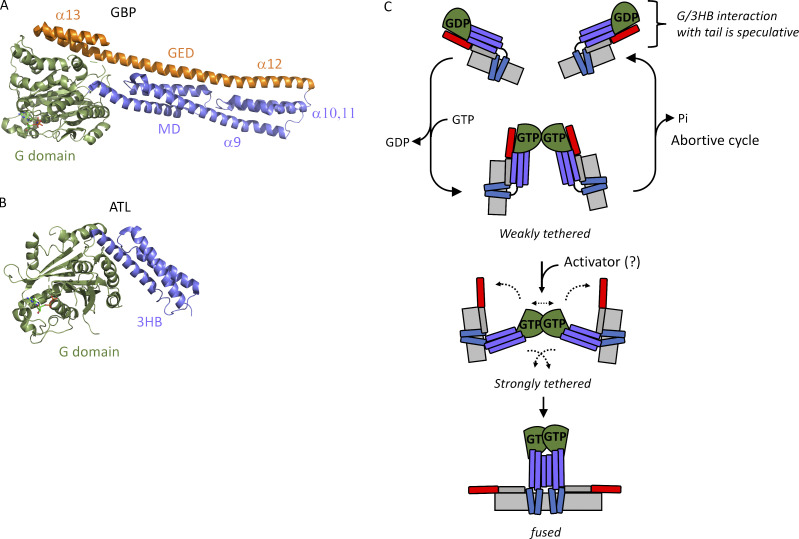
ER network formation depends on membrane fusion by the atlastin (ATL) GTPase. In humans, three paralogs are differentially expressed with divergent N- and C-terminal extensions, but their respective roles remain unknown. This is partly because, unlike Drosophila ATL, the fusion activity of human ATLs has not been reconstituted. Here, we report successful reconstitution of fusion activity by the human ATLs. Unexpectedly, the major splice isoforms of ATL1 and ATL2 are each autoinhibited, albeit to differing degrees. For the more strongly inhibited ATL2, autoinhibition mapped to a C-terminal α-helix is predicted to be continuous with an amphipathic helix required for fusion. Charge reversal of residues in the inhibitory domain strongly activated its fusion activity, and overexpression of this disinhibited version caused ER collapse. Neurons express an ATL2 splice isoform whose sequence differs in the inhibitory domain, and this form showed full fusion activity. These findings reveal autoinhibition and alternate splicing as regulators of atlastin-mediated ER fusion.
© 2021 Crosby et al.
Figures
Similar articles
-
The Atlastin C-terminal tail is an amphipathic helix that perturbs the bilayer structure during endoplasmic reticulum homotypic fusion.J Biol Chem. 2015 Feb 20;290(8):4772-4783. doi: 10.1074/jbc.M114.601823. Epub 2015 Jan 2. J Biol Chem. 2015. PMID: 25555915 Free PMC article.
-
Comparison of human and Drosophila atlastin GTPases.Protein Cell. 2015 Feb;6(2):139-46. doi: 10.1007/s13238-014-0118-0. Epub 2014 Nov 20. Protein Cell. 2015. PMID: 25407413 Free PMC article.
-
Reciprocal regulation between lunapark and atlastin facilitates ER three-way junction formation.Protein Cell. 2019 Jul;10(7):510-525. doi: 10.1007/s13238-018-0595-7. Epub 2018 Nov 29. Protein Cell. 2019. PMID: 30498943 Free PMC article.
-
Connecting tubules: mechanisms of endoplasmic reticulum membrane fusion.Biochem Soc Trans. 2025 Jun 30;53(3):699-707. doi: 10.1042/BST20253043. Biochem Soc Trans. 2025. PMID: 40587263 Free PMC article. Review.
-
Reconstituting the reticular ER network - mechanistic implications and open questions.J Cell Sci. 2019 Jan 22;132(4):jcs227611. doi: 10.1242/jcs.227611. J Cell Sci. 2019. PMID: 30670475 Review.
Cited by
-
Structural and functional characterization of the brain-specific dynamin superfamily member RNF112.Proc Natl Acad Sci U S A. 2025 Apr 15;122(15):e2419449122. doi: 10.1073/pnas.2419449122. Epub 2025 Apr 8. Proc Natl Acad Sci U S A. 2025. PMID: 40198702 Free PMC article.
-
Allosteric control of dynamin-related protein 1-catalyzed mitochondrial fission through a conserved disordered C-terminal Short Linear Motif.Res Sq [Preprint]. 2023 Jul 18:rs.3.rs-3161608. doi: 10.21203/rs.3.rs-3161608/v2. Res Sq. 2023. PMID: 37503116 Free PMC article. Preprint.
-
The atlastin paralogs: The complexity in the tails.J Cell Biol. 2023 Jul 3;222(7):e202305116. doi: 10.1083/jcb.202305116. Epub 2023 Jun 16. J Cell Biol. 2023. PMID: 37327452 Free PMC article.
-
High Atlastin 2-2 (ATL2-2) Expression Associates with Worse Prognosis in Estrogen-Receptor-Positive Breast Cancer.Genes (Basel). 2023 Jul 29;14(8):1559. doi: 10.3390/genes14081559. Genes (Basel). 2023. PMID: 37628611 Free PMC article.
-
Human atlastin-3 is a constitutive ER membrane fusion catalyst.J Cell Biol. 2023 Jul 3;222(7):e202211021. doi: 10.1083/jcb.202211021. Epub 2023 Apr 26. J Cell Biol. 2023. PMID: 37102997 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
