

Ligands binding to the prion protein induce its proteolytic release with therapeutic potential in neurodegenerative proteinopathies
- PMID: 34818048
- PMCID: PMC8612689
- DOI: 10.1126/sciadv.abj1826
Ligands binding to the prion protein induce its proteolytic release with therapeutic potential in neurodegenerative proteinopathies
Abstract
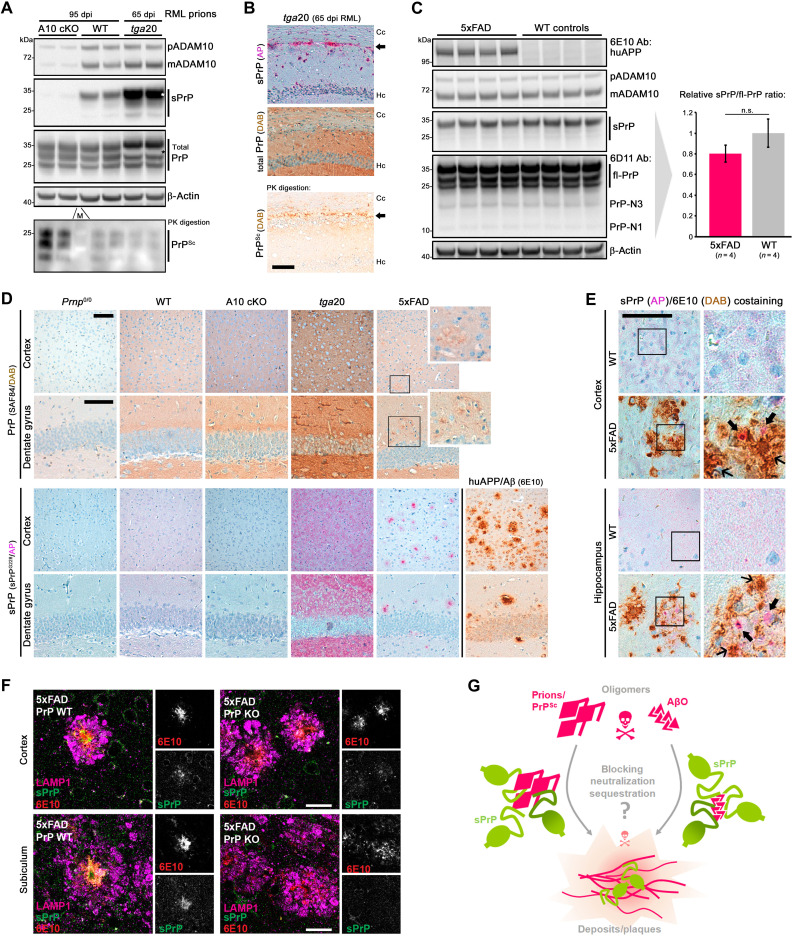
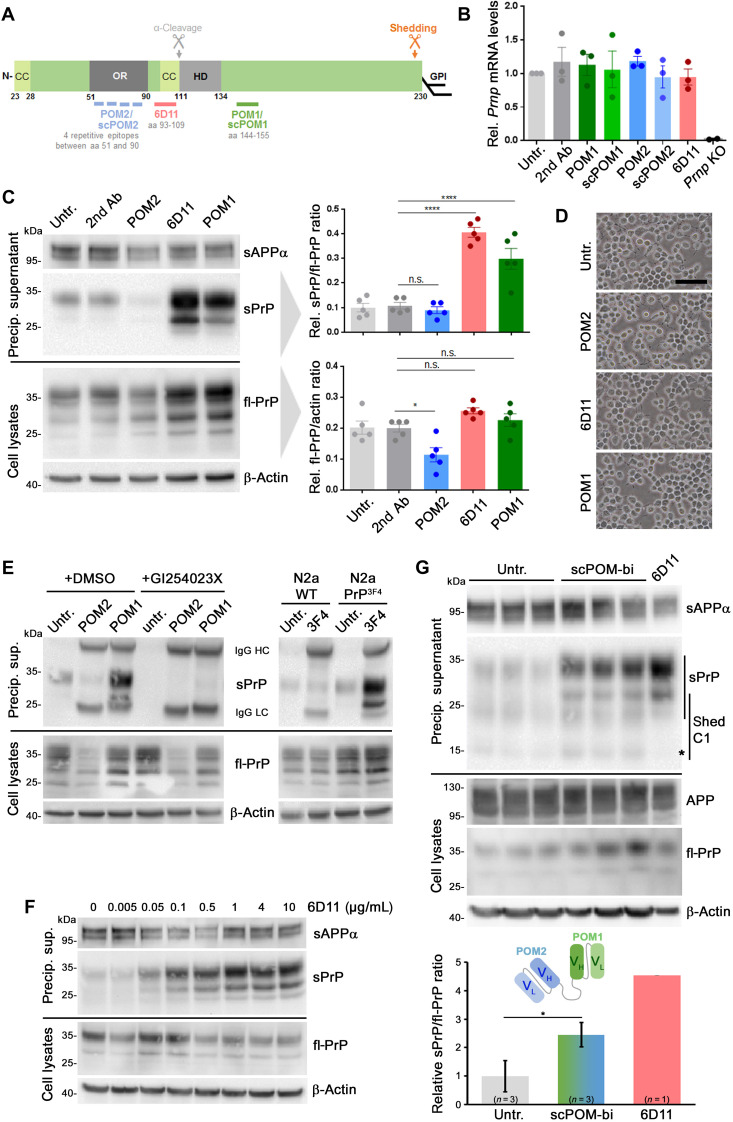
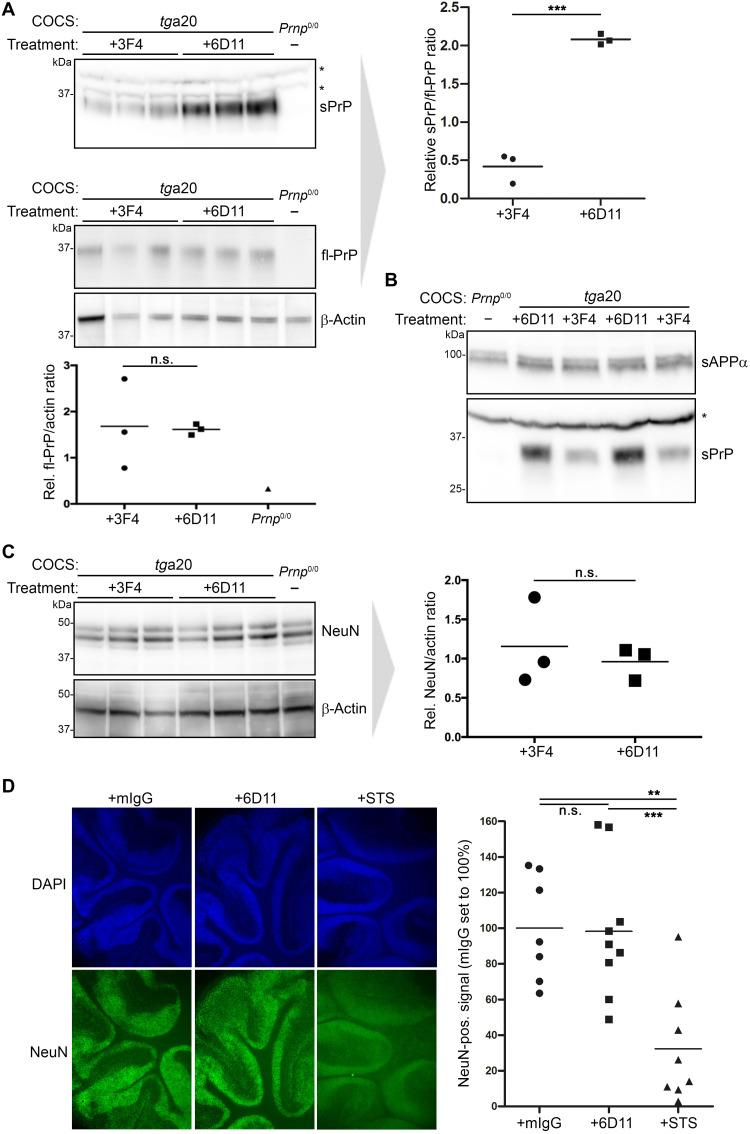
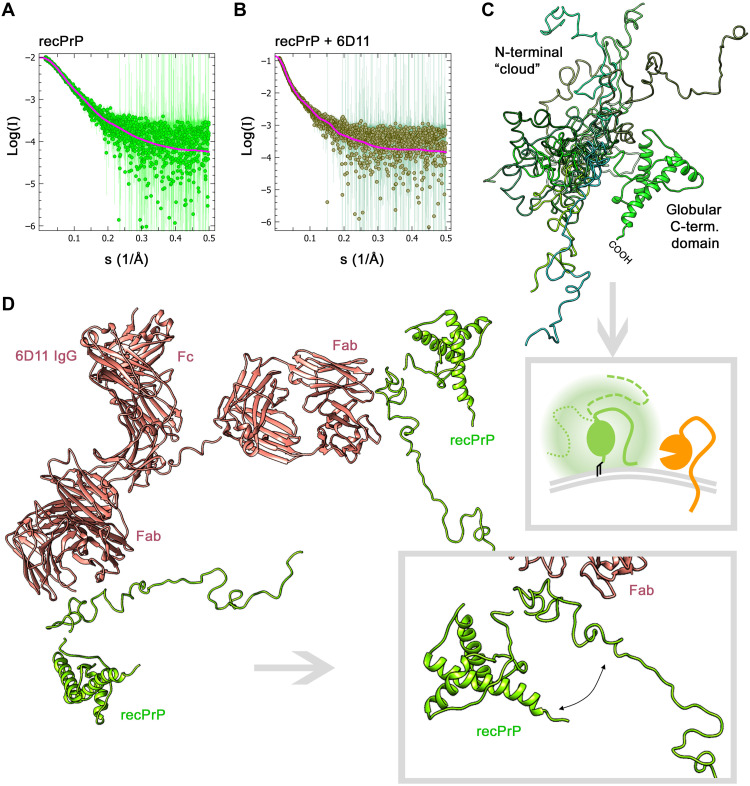
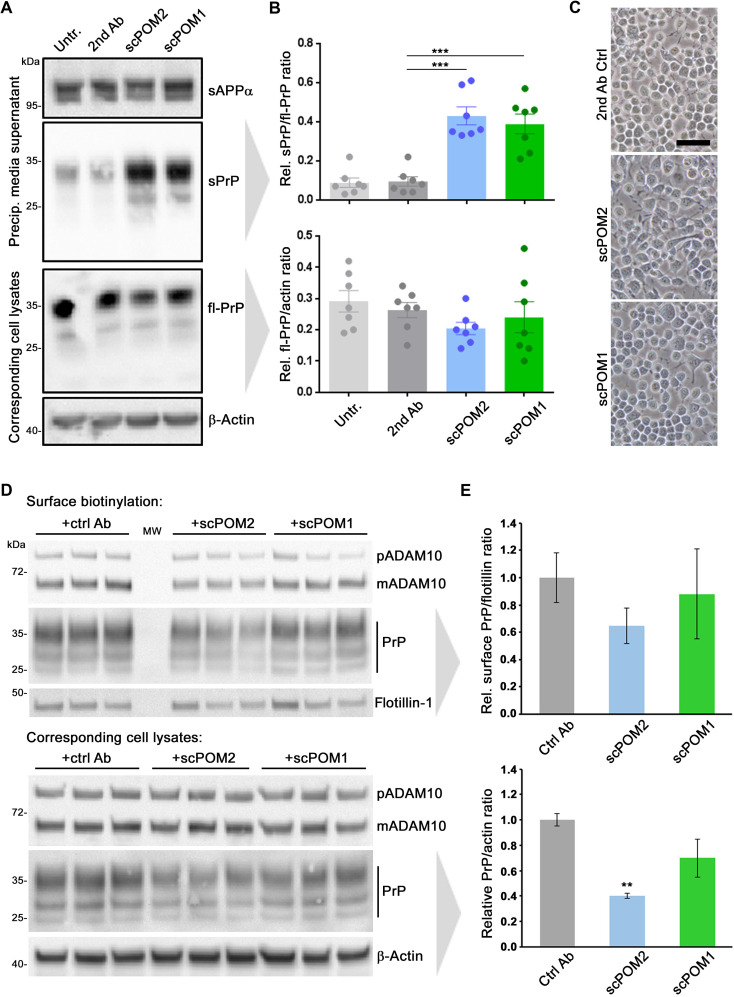
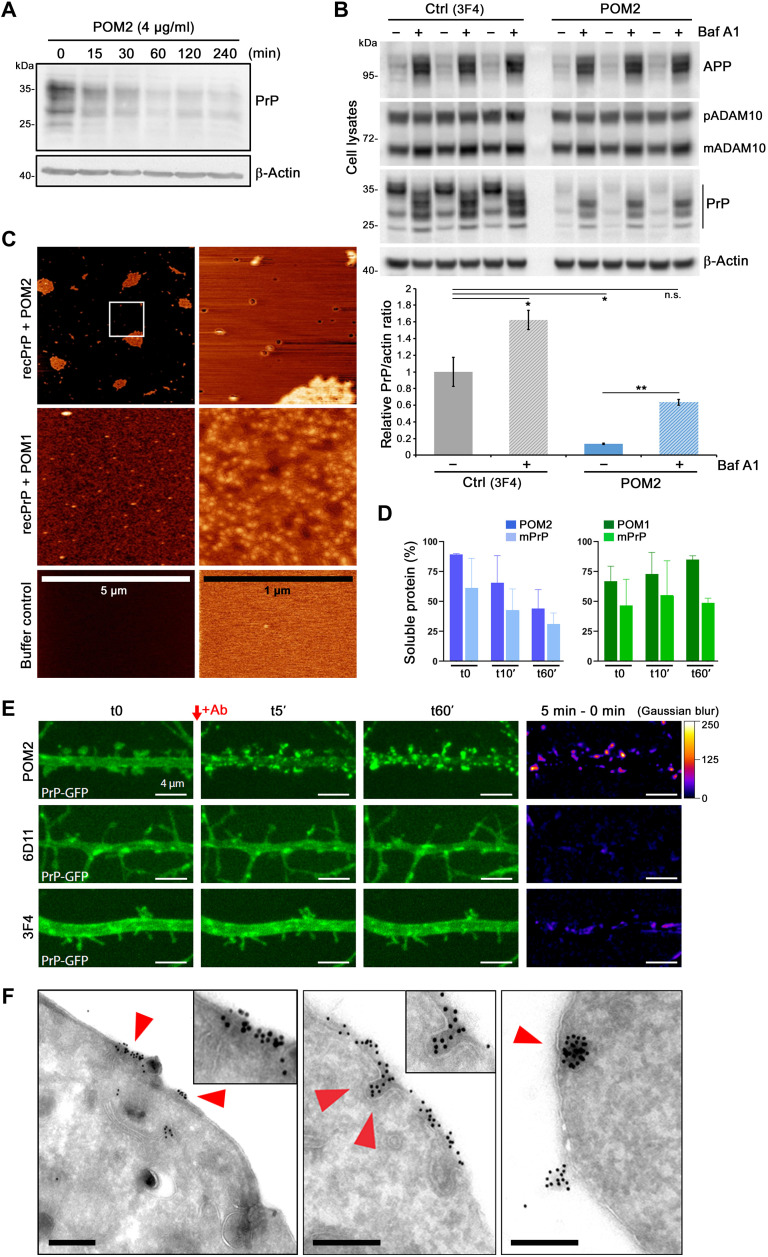
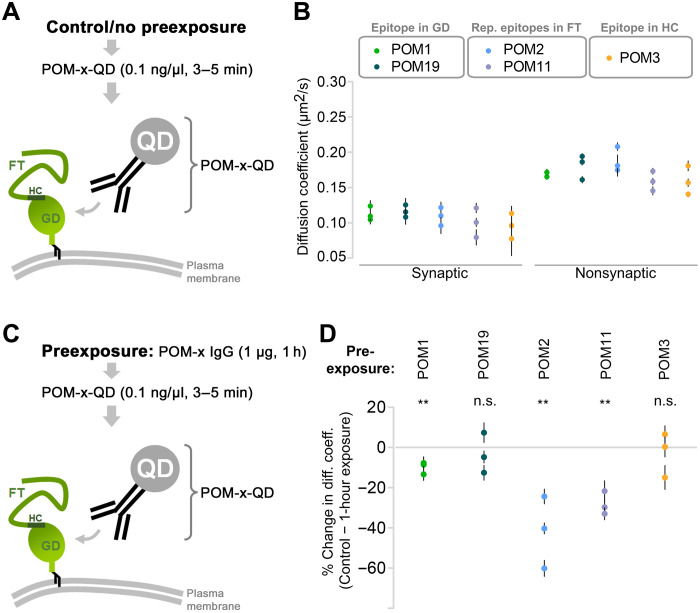
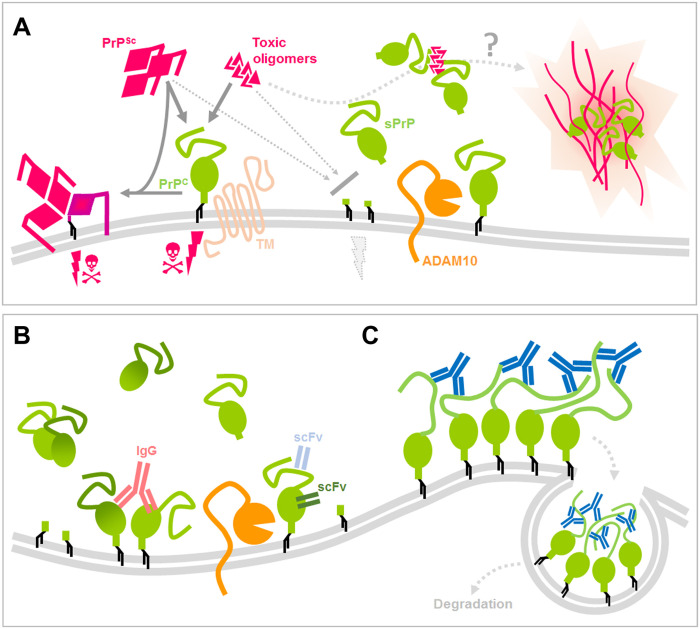
The prion protein (PrPC) is a central player in neurodegenerative diseases, such as prion diseases or Alzheimer’s disease. In contrast to disease-promoting cell surface PrPC, extracellular fragments act neuroprotective by blocking neurotoxic disease-associated protein conformers. Fittingly, PrPC release by the metalloprotease ADAM10 represents a protective mechanism. We used biochemical, cell biological, morphological, and structural methods to investigate mechanisms stimulating this proteolytic shedding. Shed PrP negatively correlates with prion conversion and is markedly redistributed in murine brain in the presence of prion deposits or amyloid plaques, indicating a sequestrating activity. PrP-directed ligands cause structural changes in PrPC and increased shedding in cells and organotypic brain slice cultures. As an exception, some PrP-directed antibodies targeting repetitive epitopes do not cause shedding but surface clustering, endocytosis, and degradation of PrPC. Both mechanisms may contribute to beneficial actions described for PrP-directed ligands and pave the way for new therapeutic strategies against currently incurable neurodegenerative diseases.
Figures
References
-
- Aguzzi A., Haass C., Games played by rogue proteins in prion disorders and Alzheimer’s disease. Science 302, 814–818 (2003). - PubMed
-
- Vaquer-Alicea J., Diamond M. I., Propagation of protein aggregation in neurodegenerative diseases. Annu. Rev. Biochem. 88, 785–810 (2019). - PubMed
-
- Checler F., Vincent B., Alzheimer’s and prion diseases: Distinct pathologies, common proteolytic denominators. Trends Neurosci. 25, 616–620 (2002). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
