

Interspecies variation in hominid gut microbiota controls host gene regulation
- PMID: 34818542
- PMCID: PMC8647622
- DOI: 10.1016/j.celrep.2021.110057
Interspecies variation in hominid gut microbiota controls host gene regulation
Abstract
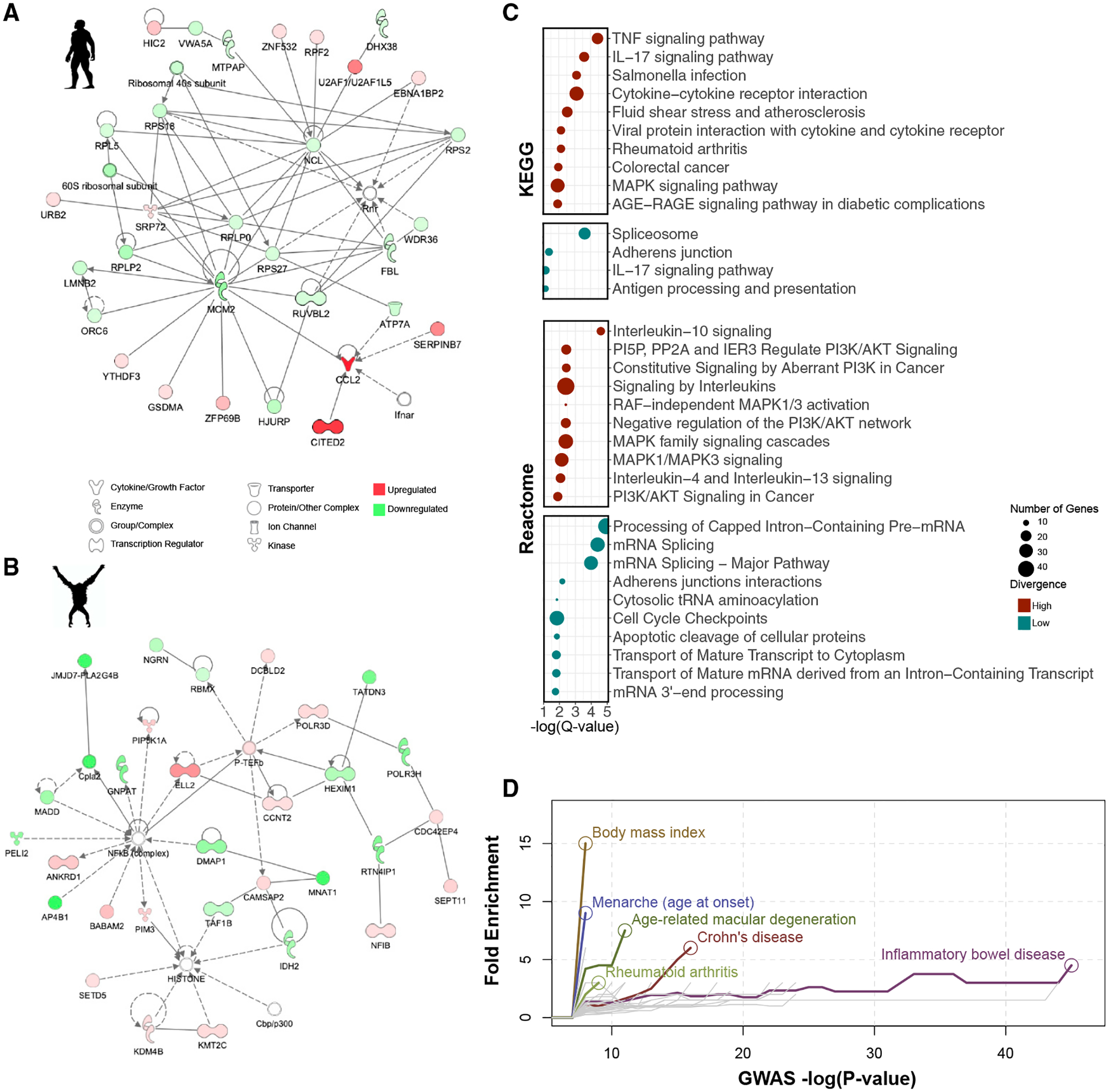
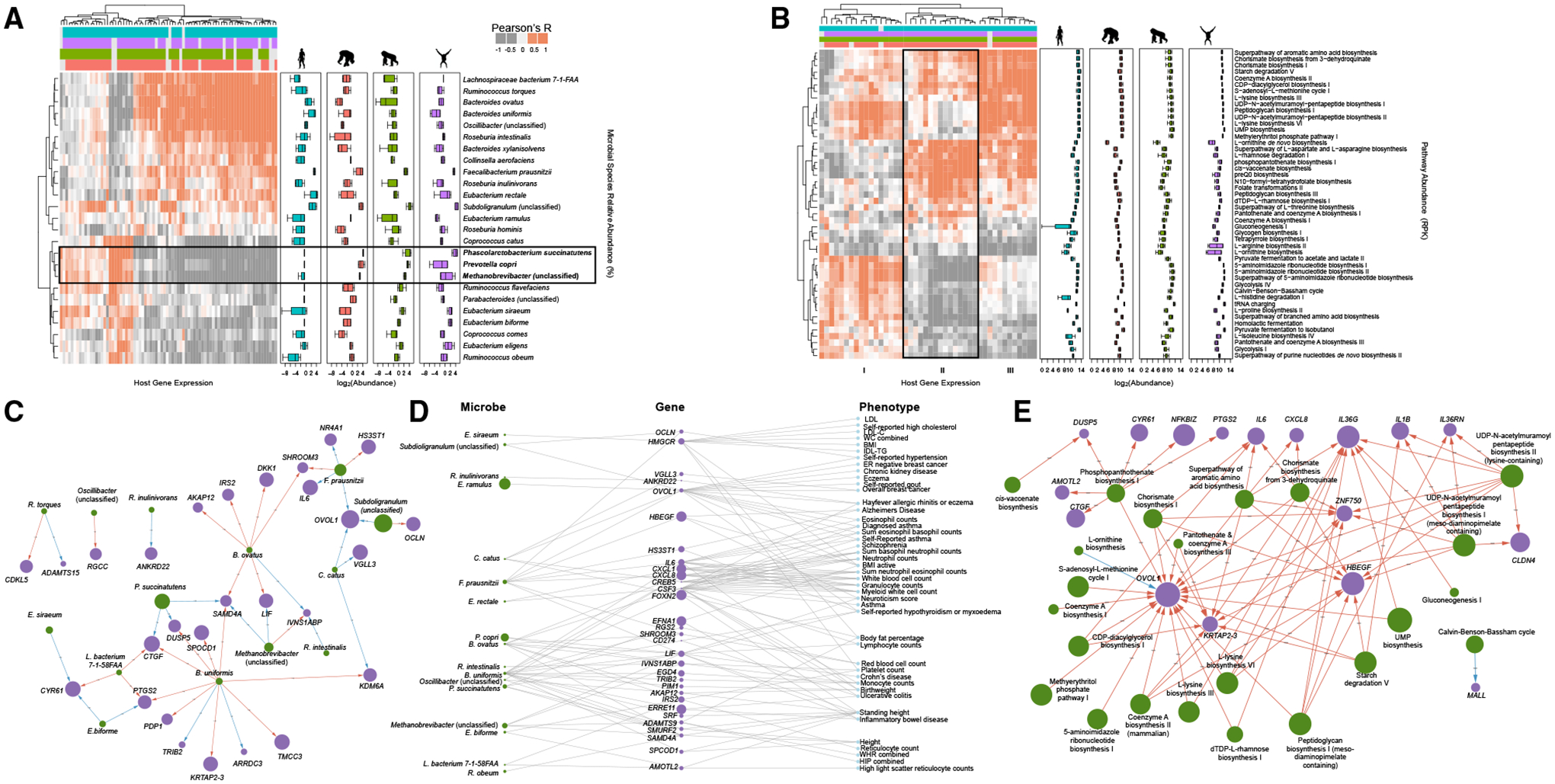
The gut microbiome exhibits extreme compositional variation between hominid hosts. However, it is unclear how this variation impacts host physiology across species and whether this effect can be mediated through microbial regulation of host gene expression in interacting epithelial cells. Here, we characterize the transcriptional response of human colonic epithelial cells in vitro to live microbial communities extracted from humans, chimpanzees, gorillas, and orangutans. We find that most host genes exhibit a conserved response, whereby they respond similarly to the four hominid microbiomes. However, hundreds of host genes exhibit a divergent response, whereby they respond only to microbiomes from specific host species. Such genes are associated with intestinal diseases in humans, including inflammatory bowel disease and Crohn's disease. Last, we find that inflammation-associated microbial species regulate the expression of host genes previously associated with inflammatory bowel disease, suggesting health-related consequences for species-specific host-microbiome interactions across hominids.
Keywords: Gut microbiome, Primates, Hominids, Gene regulation.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
