

Compensatory role of endogenous sulfur dioxide in nitric oxide deficiency-induced hypertension
- PMID: 34818607
- PMCID: PMC8626683
- DOI: 10.1016/j.redox.2021.102192
Compensatory role of endogenous sulfur dioxide in nitric oxide deficiency-induced hypertension
Abstract
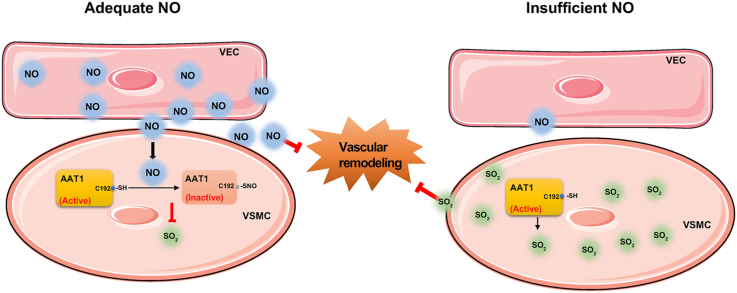
Objective: This study aimed to determine the communicational pattern of gaseous signaling molecules sulfur dioxide (SO2) and nitric oxide (NO) between vascular endothelial cells (VECs) and vascular smooth muscle cells (VSMCs), and elucidate the compensatory role and significance of endogenous SO2 in the development of hypertension due to NO deficiency.
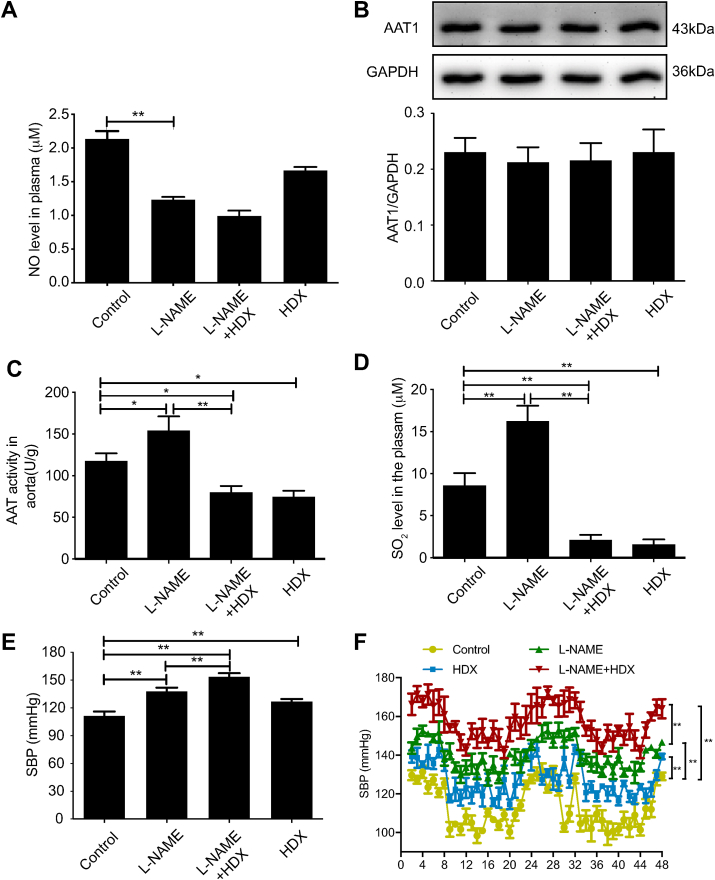
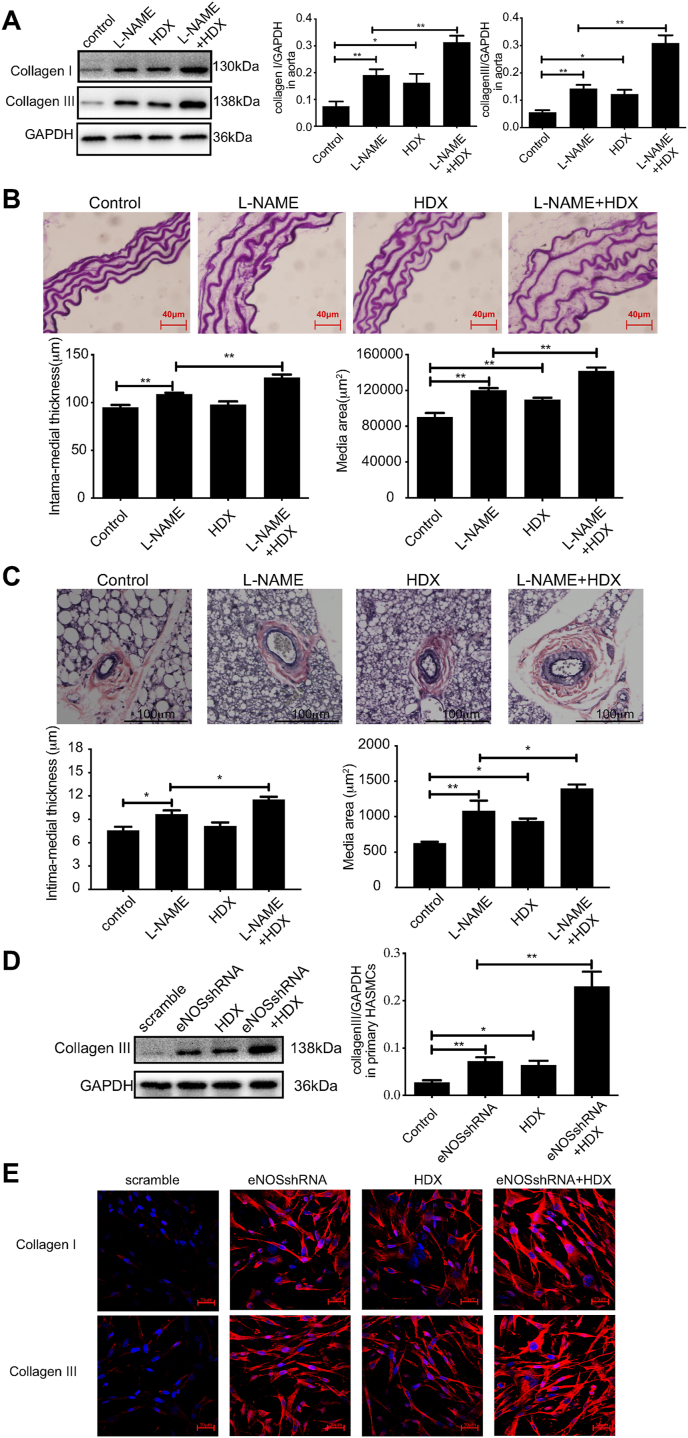
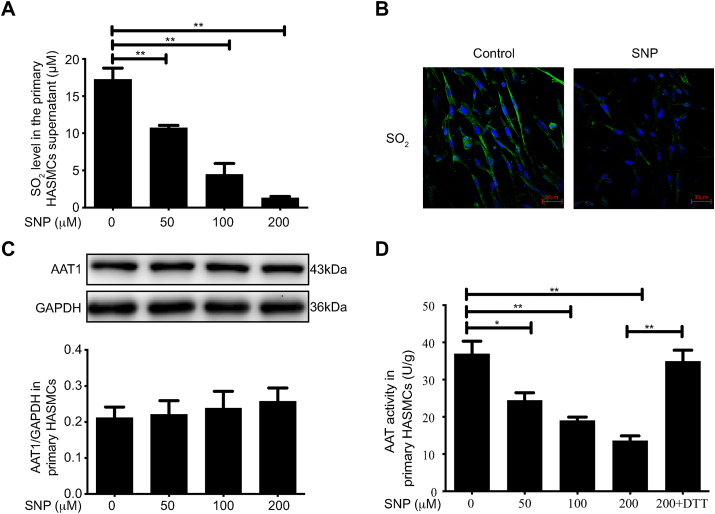
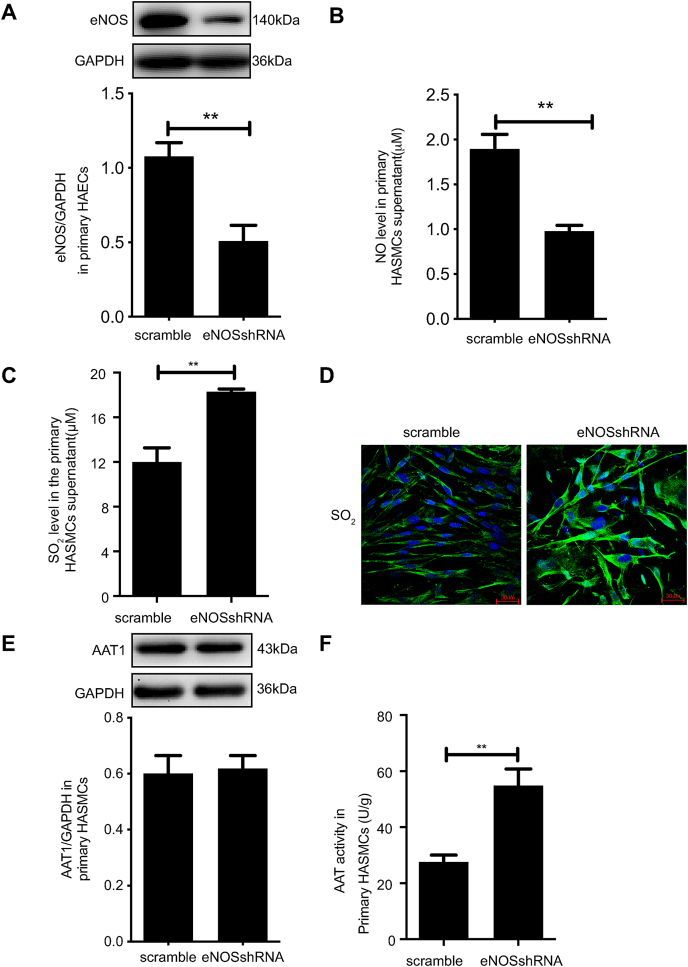
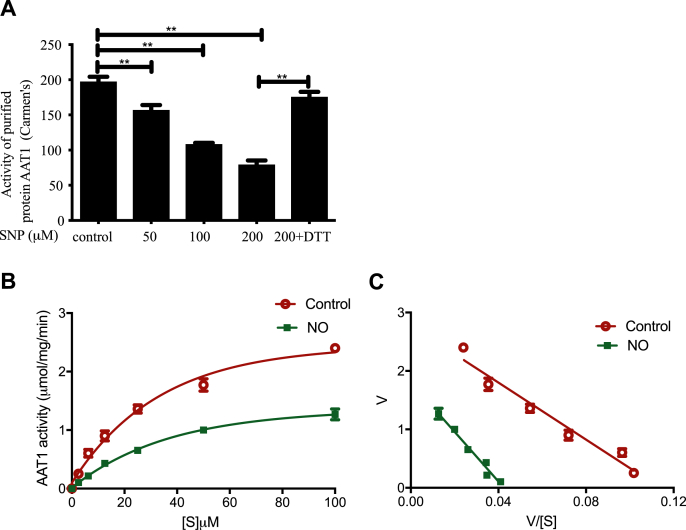
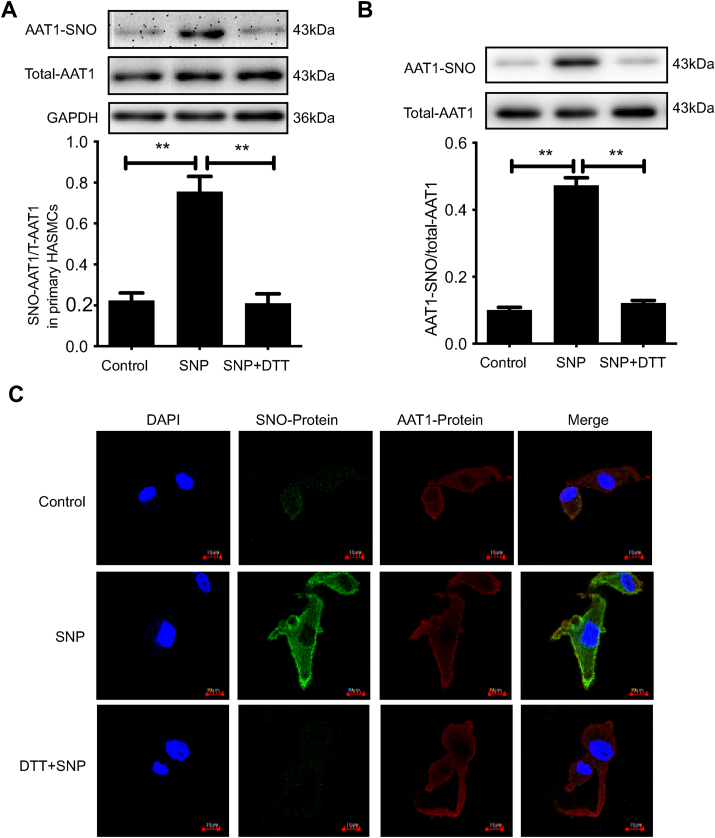
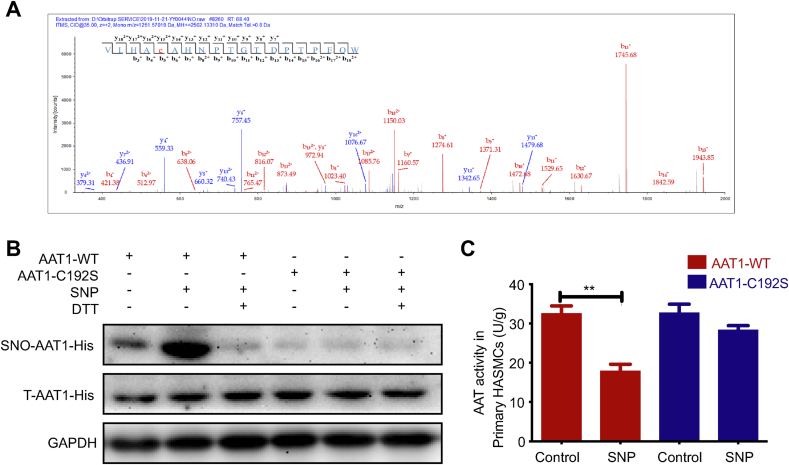
Approach and results: Blood pressure was monitored by the tail-cuff and implantable physiological signal telemetry in L-nitro-arginine methyl ester (l-NAME)-induced hypertensive mice, and structural alterations of mouse aortic vessels were detected by the elastic fiber staining method. l-NAME-treated mice showed decreased plasma NO levels, increased SO2 levels, vascular remodeling, and increased blood pressure, and application of l-aspartate-β-hydroxamate, which inhibits SO2 production, further aggravated vascular structural remodeling and increased blood pressure. Moreover, in a co-culture system of HAECs and HASMCs, NO from HAECs did not influence aspartate aminotransferase (AAT)1 protein expression but decreased AAT1 activity in HASMCs, thereby resulting in the inhibition of endogenous SO2 production. Furthermore, NO promoted S-nitrosylation of AAT1 protein in HASMCs and purified AAT1 protein. Liquid chromatography with tandem mass spectrometry showed that the Cys192 site of AAT1 purified protein was modified by S-nitrosylation. In contrast, dithiothreitol or C192S mutations in HASMCs blocked NO-induced AAT1 S-nitrosylation and restored AAT1 enzyme activity.
Conclusion: Endothelium-derived NO inhibits AAT activity by nitrosylating AAT1 at the Cys192 site and reduces SO2 production in HASMCs. Our findings suggest that SO2 acts as a compensatory defense system to antagonize vascular structural remodeling and hypertension when the endogenous NO pathway is disturbed.
Keywords: Aspartate aminotransferase; Endothelial cells; Remodeling; S-Nitrosylation; Sulfur dioxide.
Copyright © 2021 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Kearney P.M., Whelton M., Reynolds K., Muntner P., Whelton P.K., He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–223. - PubMed
-
- Rapoport R.M., Draznin M.B., Murad F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature. 1983;306:174–176. - PubMed
-
- Forstermann U., Mulsch A., Bohme E., Busse R. Stimulation of soluble guanylate cyclase by an acetylcholine-induced endothelium-derived factor from rabbit and canine arteries. Circ. Res. 1986;58:531–538. - PubMed
-
- Ignarro L.J., Harbison R.G., Wood K.S., Kadowitz P.J. Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J. Pharmacol. Exp. Therapeut. 1986;237:893–900. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
