

Global phylogeny of Treponema pallidum lineages reveals recent expansion and spread of contemporary syphilis
- PMID: 34819643
- PMCID: PMC8612932
- DOI: 10.1038/s41564-021-01000-z
Global phylogeny of Treponema pallidum lineages reveals recent expansion and spread of contemporary syphilis
Abstract
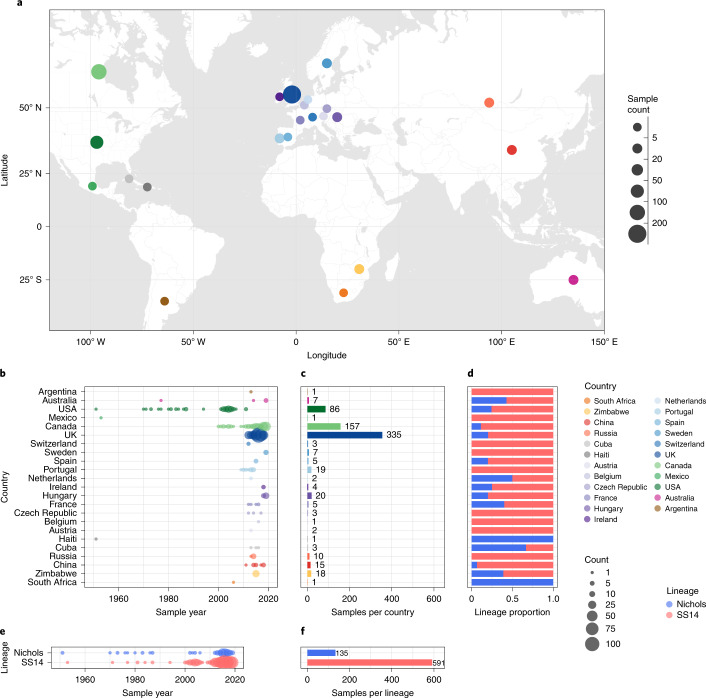
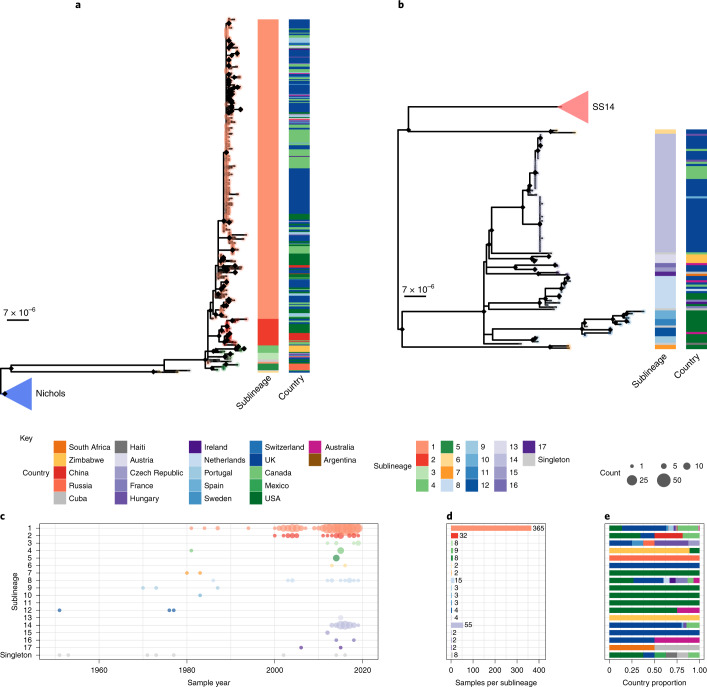
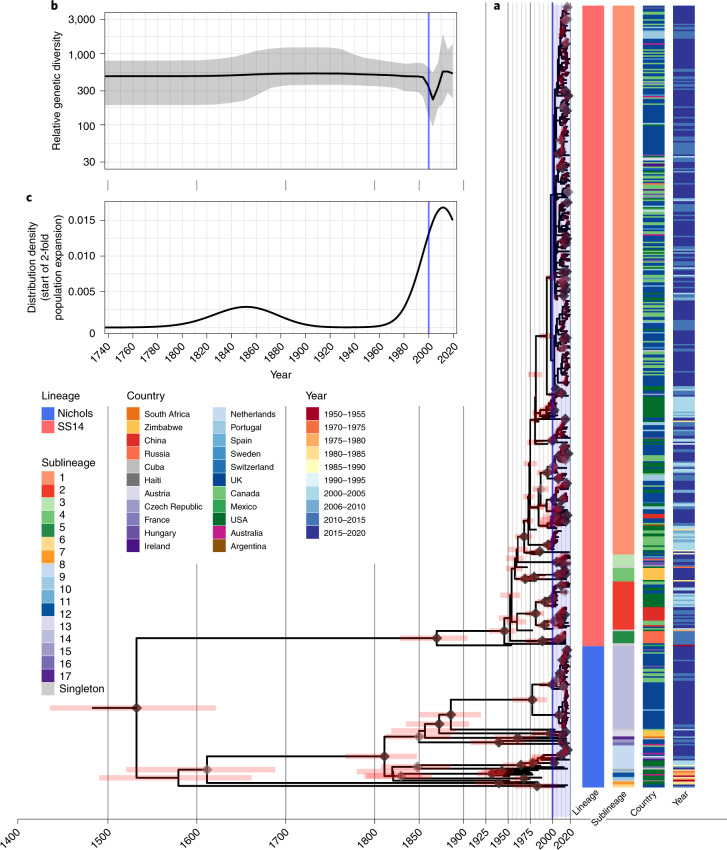
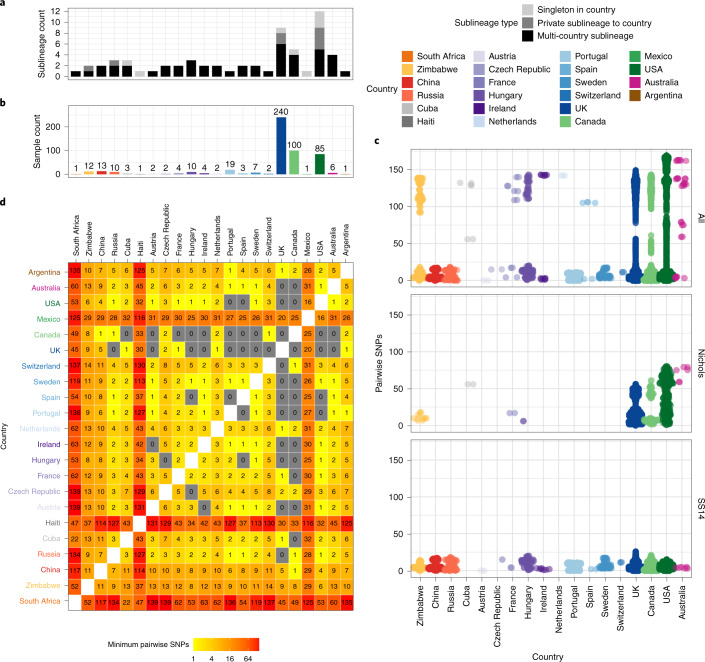
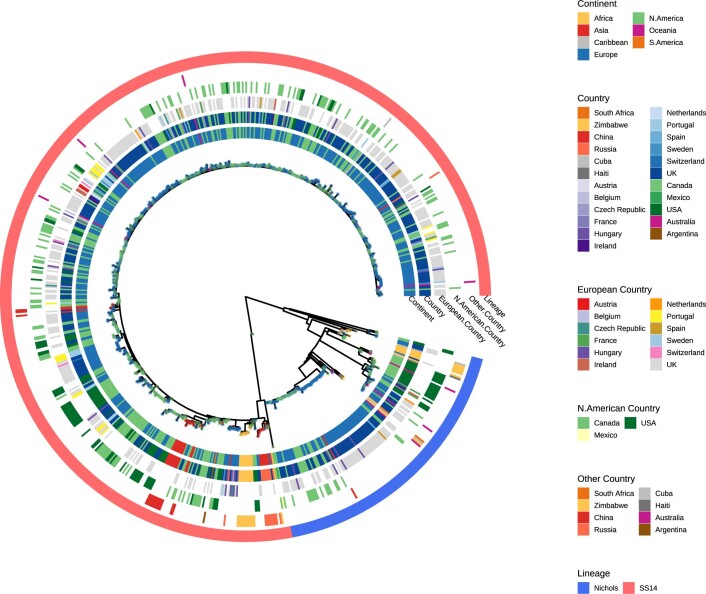
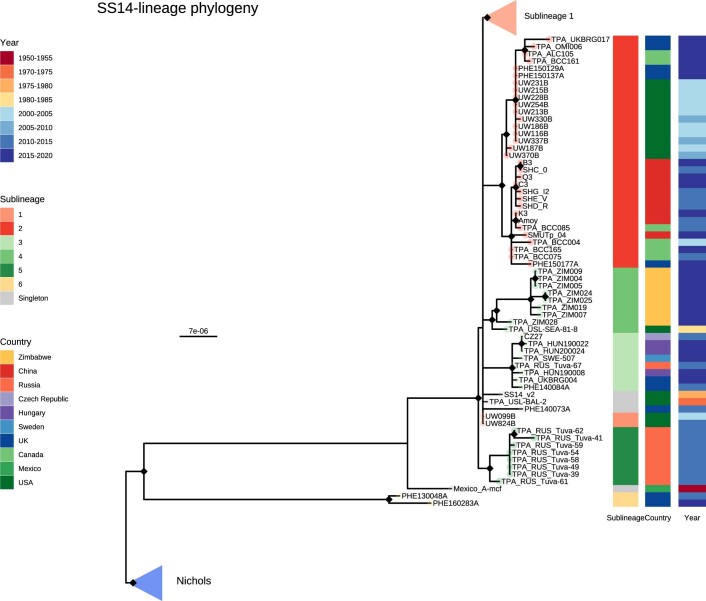
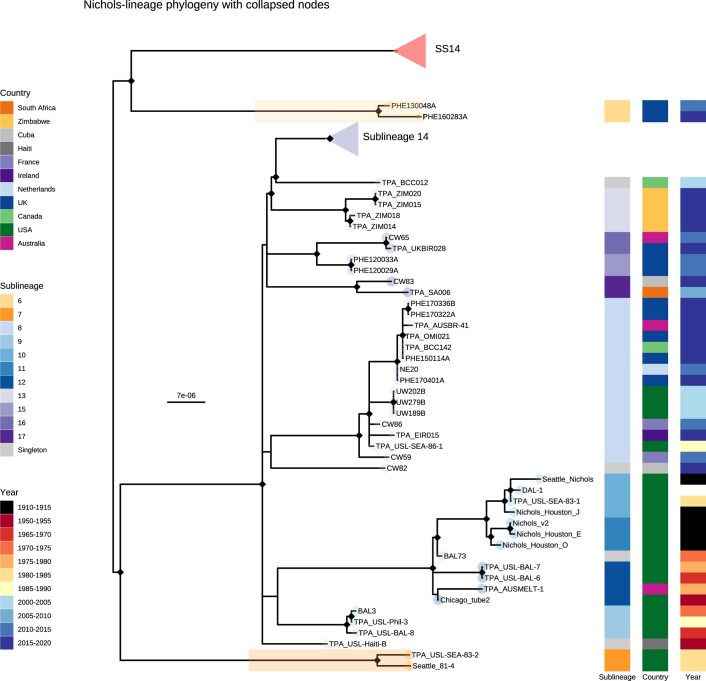
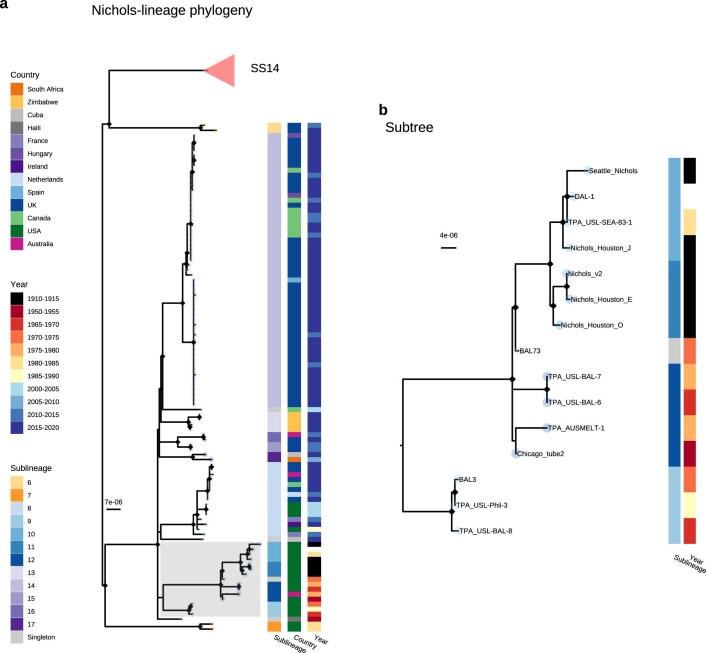
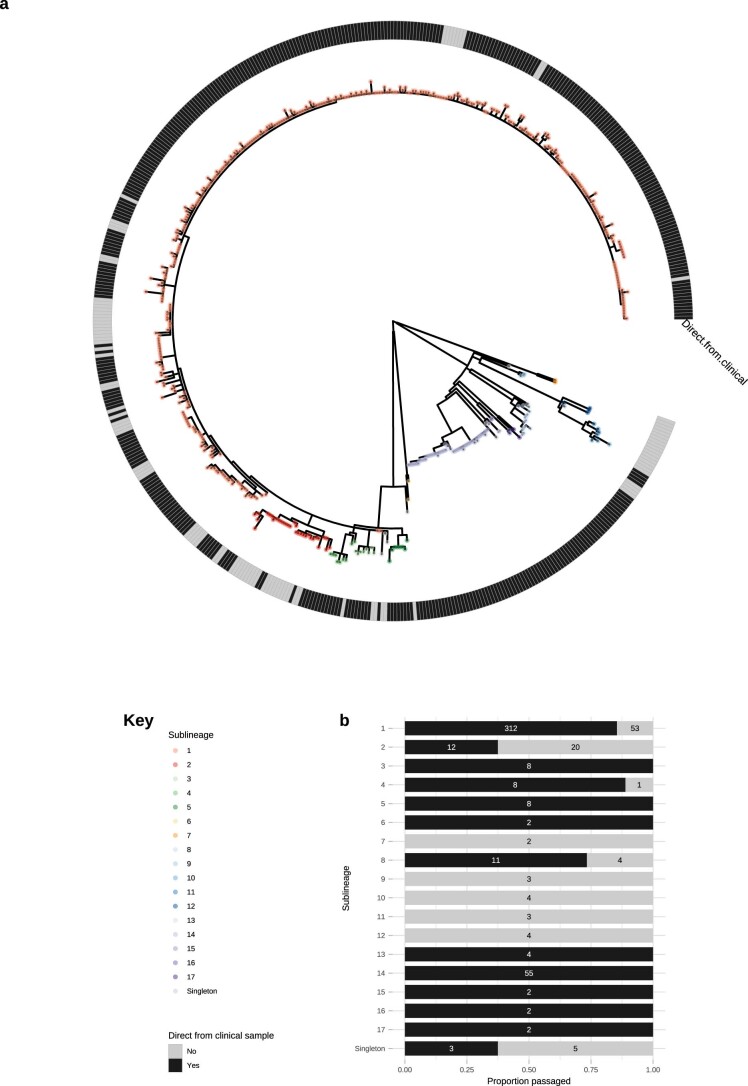
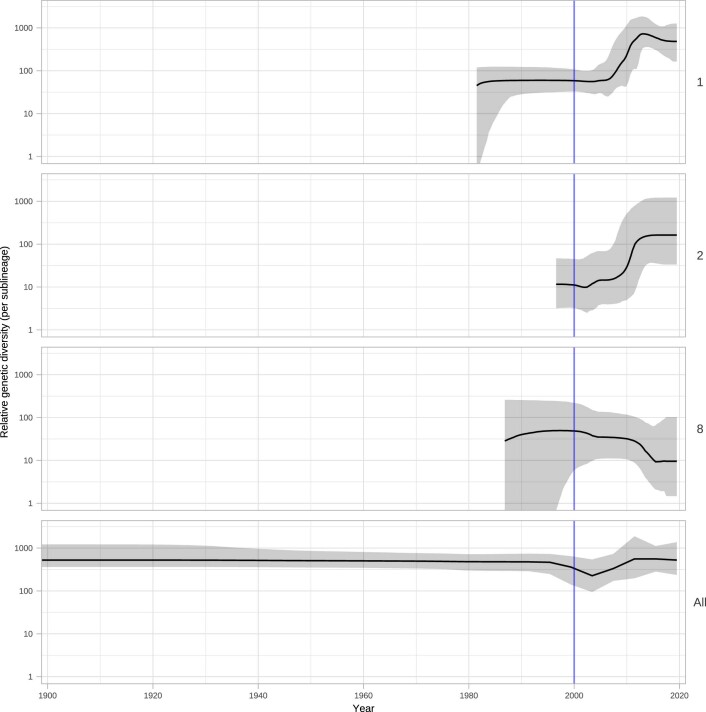
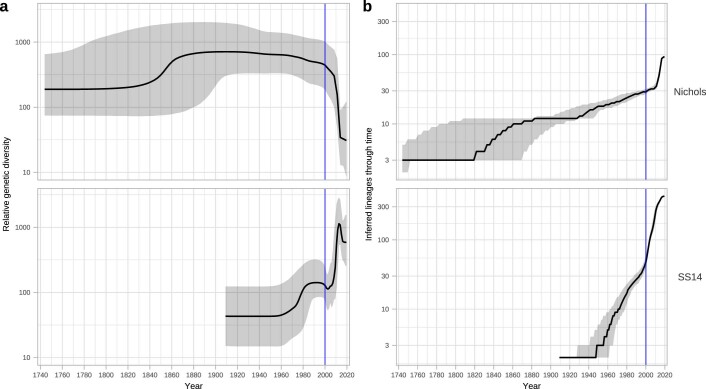
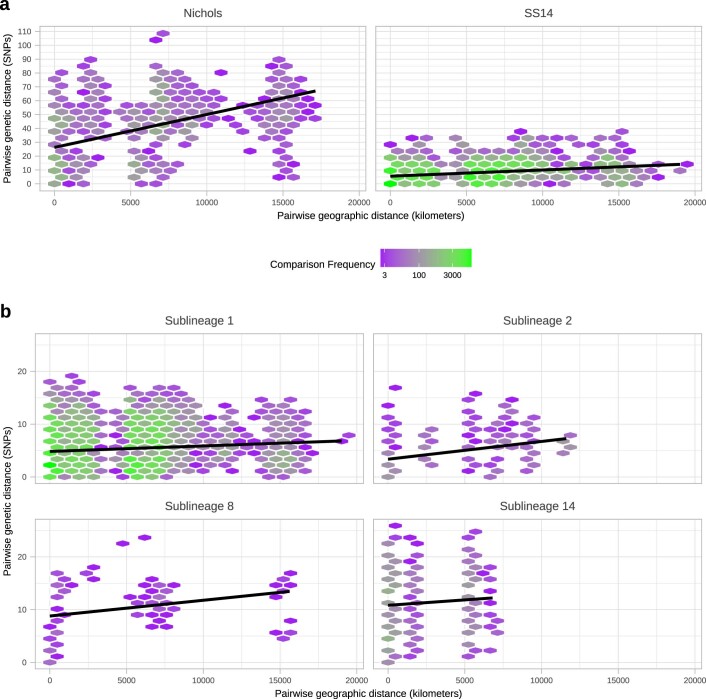
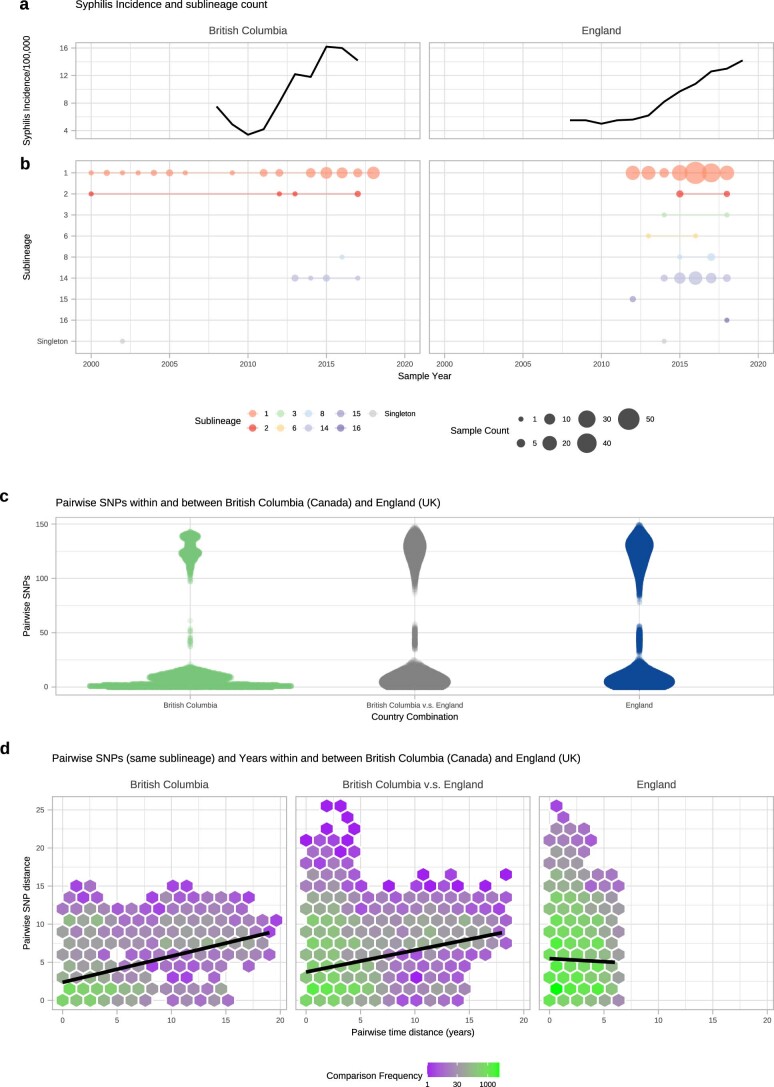
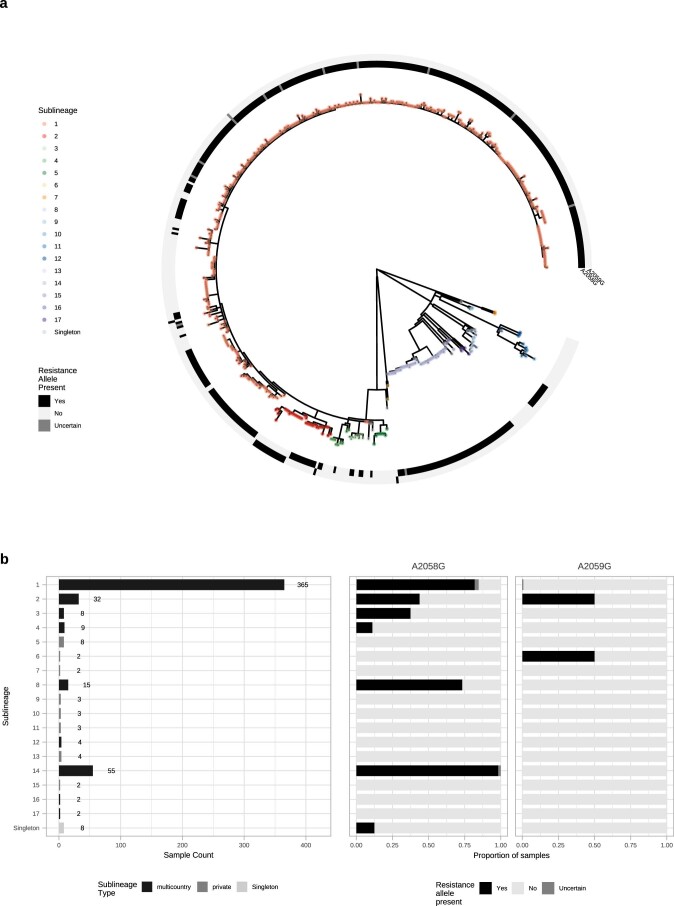
Syphilis, which is caused by the sexually transmitted bacterium Treponema pallidum subsp. pallidum, has an estimated 6.3 million cases worldwide per annum. In the past ten years, the incidence of syphilis has increased by more than 150% in some high-income countries, but the evolution and epidemiology of the epidemic are poorly understood. To characterize the global population structure of T. pallidum, we assembled a geographically and temporally diverse collection of 726 genomes from 626 clinical and 100 laboratory samples collected in 23 countries. We applied phylogenetic analyses and clustering, and found that the global syphilis population comprises just two deeply branching lineages, Nichols and SS14. Both lineages are currently circulating in 12 of the 23 countries sampled. We subdivided T. p. pallidum into 17 distinct sublineages to provide further phylodynamic resolution. Importantly, two Nichols sublineages have expanded clonally across 9 countries contemporaneously with SS14. Moreover, pairwise genome analyses revealed examples of isolates collected within the last 20 years from 14 different countries that had genetically identical core genomes, which might indicate frequent exchange through international transmission. It is striking that most samples collected before 1983 are phylogenetically distinct from more recently isolated sublineages. Using Bayesian temporal analysis, we detected a population bottleneck occurring during the late 1990s, followed by rapid population expansion in the 2000s that was driven by the dominant T. pallidum sublineages circulating today. This expansion may be linked to changing epidemiology, immune evasion or fitness under antimicrobial selection pressure, since many of the contemporary syphilis lineages we have characterized are resistant to macrolides.
© 2021. The Author(s).
Conflict of interest statement
M.K. declares institutional funding from Roche, Hologic and Siemens, which is unrelated to this work. The remaining authors declare no competing interests. The funders had no input into the study design, interpretation or decision to submit for publication.
Figures
Comment in
-
Bottleneck and spread of Treponema pallidum.Nat Rev Microbiol. 2022 Feb;20(2):64. doi: 10.1038/s41579-021-00673-x. Nat Rev Microbiol. 2022. PMID: 34880455 No abstract available.
References
-
- McGough, L. J. & Erbelding, E. in Pathogenic Treponema: Molecular and Cellular Biology (eds Radolf, J. D. & Lukehart, S. A.) 183–195 (Caister Academic Press, 2006).
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
