

Signaling pathways and intervention therapies in sepsis
- PMID: 34824200
- PMCID: PMC8613465
- DOI: 10.1038/s41392-021-00816-9
Signaling pathways and intervention therapies in sepsis
Abstract
Sepsis is defined as life-threatening organ dysfunction caused by dysregulated host systemic inflammatory and immune response to infection. Over decades, advanced understanding of host-microorganism interaction has gradually unmasked the genuine nature of sepsis, guiding toward new definition and novel therapeutic approaches. Diverse clinical manifestations and outcomes among infectious patients have suggested the heterogeneity of immunopathology, while systemic inflammatory responses and deteriorating organ function observed in critically ill patients imply the extensively hyperactivated cascades by the host defense system. From focusing on microorganism pathogenicity, research interests have turned toward the molecular basis of host responses. Though progress has been made regarding recognition and management of clinical sepsis, incidence and mortality rate remain high. Furthermore, clinical trials of therapeutics have failed to obtain promising results. As far as we know, there was no systematic review addressing sepsis-related molecular signaling pathways and intervention therapy in literature. Increasing studies have succeeded to confirm novel functions of involved signaling pathways and comment on efficacy of intervention therapies amid sepsis. However, few of these studies attempt to elucidate the underlining mechanism in progression of sepsis, while other failed to integrate preliminary findings and describe in a broader view. This review focuses on the important signaling pathways, potential molecular mechanism, and pathway-associated therapy in sepsis. Host-derived molecules interacting with activated cells possess pivotal role for sepsis pathogenesis by dynamic regulation of signaling pathways. Cross-talk and functions of these molecules are also discussed in detail. Lastly, potential novel therapeutic strategies precisely targeting on signaling pathways and molecules are mentioned.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
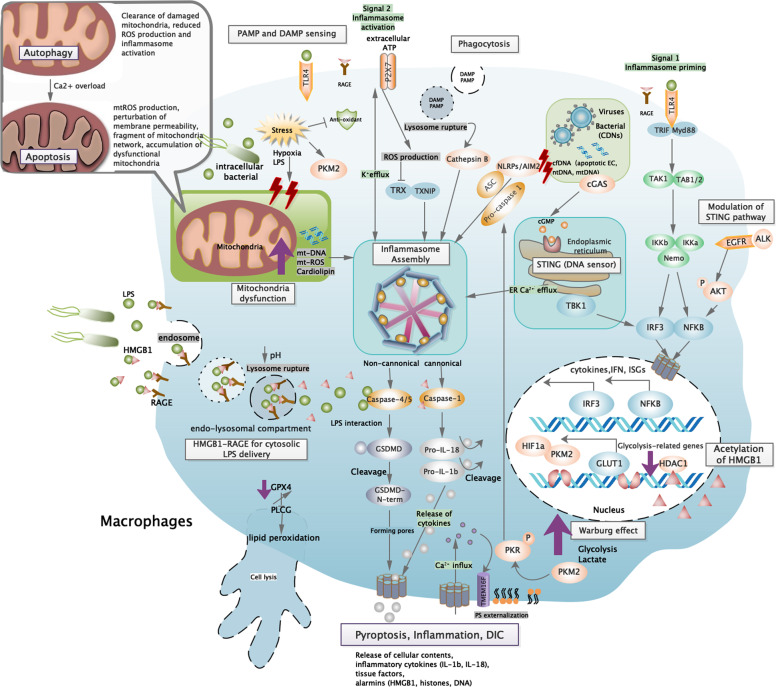
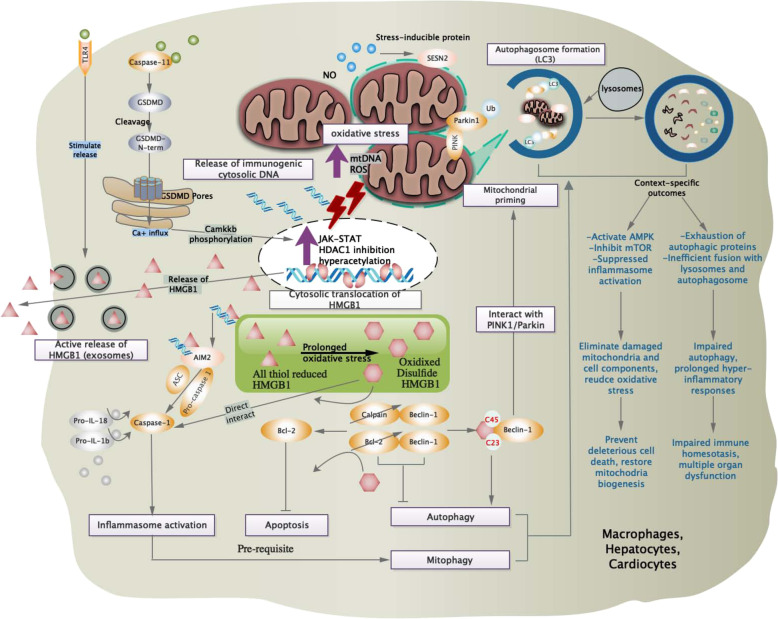
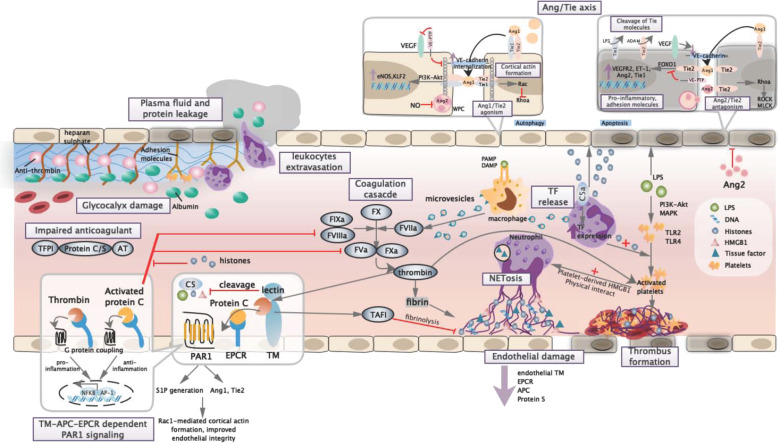
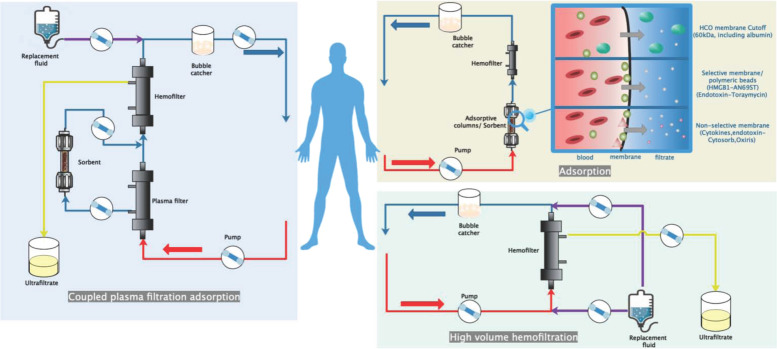
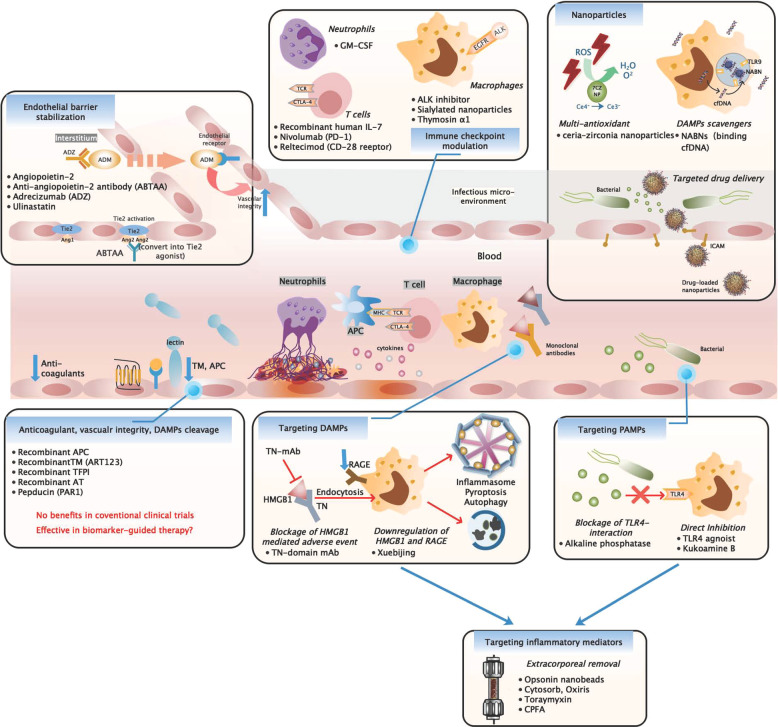
References
-
- Levy MM, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit. Care Med. 2003;31:1250–1256. - PubMed
-
- Dellinger RP, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit. Care Med. 2004;32:858–873. - PubMed
-
- Goldstein, B., Giroir, B., Randolph, A. & International Consensus Conference on Pediatric Sepsis. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr. Crit. Care Med.6, 2–8 (2005). - PubMed
-
- Davis AL, et al. American College of Critical Care Medicine clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock. Crit. Care Med. 2017;45:1061–1093. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
