

An Overview of Mitochondrial Protein Defects in Neuromuscular Diseases
- PMID: 34827632
- PMCID: PMC8615828
- DOI: 10.3390/biom11111633
An Overview of Mitochondrial Protein Defects in Neuromuscular Diseases
Abstract
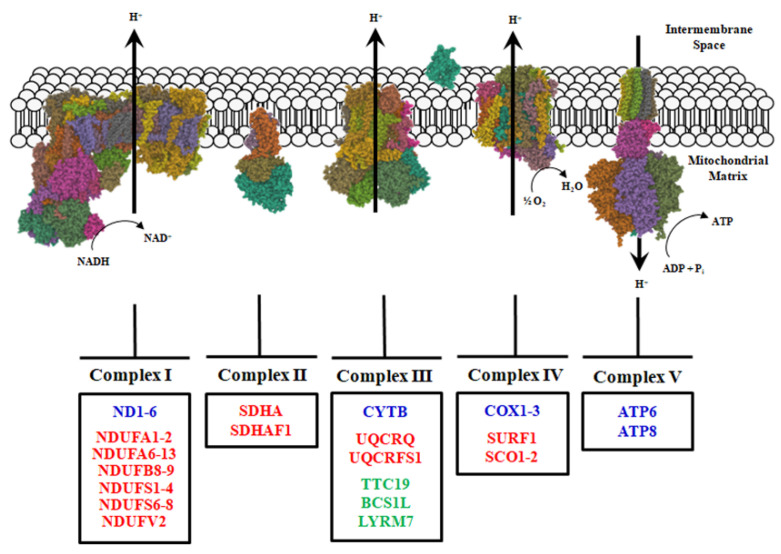
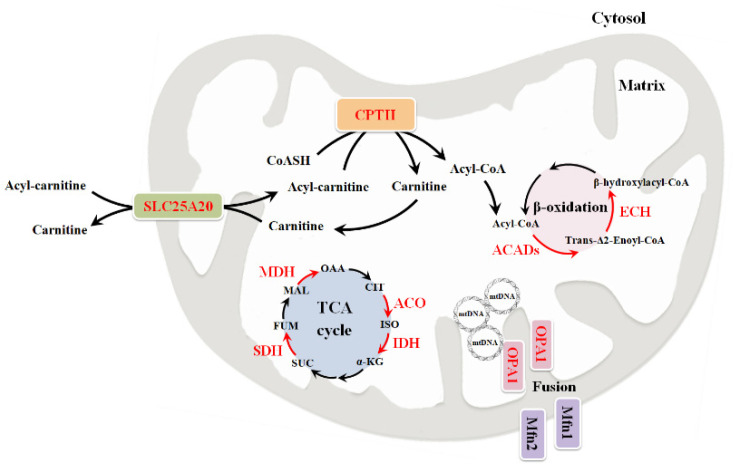
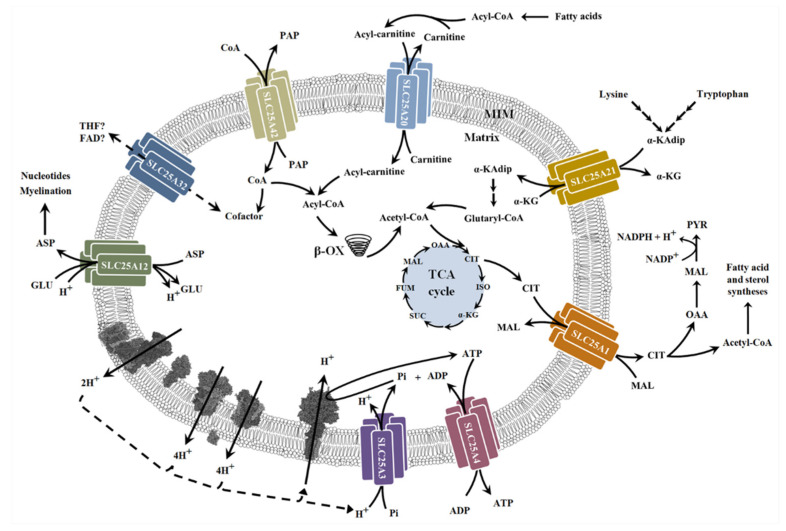
Neuromuscular diseases (NMDs) are dysfunctions that involve skeletal muscle and cause incorrect communication between the nerves and muscles. The specific causes of NMDs are not well known, but most of them are caused by genetic mutations. NMDs are generally progressive and entail muscle weakness and fatigue. Muscular impairments can differ in onset, severity, prognosis, and phenotype. A multitude of possible injury sites can make diagnosis of NMDs difficult. Mitochondria are crucial for cellular homeostasis and are involved in various metabolic pathways; for this reason, their dysfunction can lead to the development of different pathologies, including NMDs. Most NMDs due to mitochondrial dysfunction have been associated with mutations of genes involved in mitochondrial biogenesis and metabolism. This review is focused on some mitochondrial routes such as the TCA cycle, OXPHOS, and β-oxidation, recently found to be altered in NMDs. Particular attention is given to the alterations found in some genes encoding mitochondrial carriers, proteins of the inner mitochondrial membrane able to exchange metabolites between mitochondria and the cytosol. Briefly, we discuss possible strategies used to diagnose NMDs and therapies able to promote patient outcome.
Keywords: Leigh syndrome; MELAS; MERF; OXPHOS; mitochondrial carrier family; mitochondrial metabolism; myopathy; neuromuscular diseases; neuromuscular junction; therapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
