

Overcoming Microenvironment-Mediated Chemoprotection through Stromal Galectin-3 Inhibition in Acute Lymphoblastic Leukemia
- PMID: 34830047
- PMCID: PMC8624256
- DOI: 10.3390/ijms222212167
Overcoming Microenvironment-Mediated Chemoprotection through Stromal Galectin-3 Inhibition in Acute Lymphoblastic Leukemia
Abstract
Environmentally-mediated drug resistance in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) significantly contributes to relapse. Stromal cells in the bone marrow environment protect leukemia cells by secretion of chemokines as cues for BCP-ALL migration towards, and adhesion to, stroma. Stromal cells and BCP-ALL cells communicate through stromal galectin-3. Here, we investigated the significance of stromal galectin-3 to BCP-ALL cells. We used CRISPR/Cas9 genome editing to ablate galectin-3 in stromal cells and found that galectin-3 is dispensable for steady-state BCP-ALL proliferation and viability. However, efficient leukemia migration and adhesion to stromal cells are significantly dependent on stromal galectin-3. Importantly, the loss of stromal galectin-3 production sensitized BCP-ALL cells to conventional chemotherapy. We therefore tested novel carbohydrate-based small molecule compounds (Cpd14 and Cpd17) with high specificity for galectin-3. Consistent with results obtained using galectin-3-knockout stromal cells, treatment of stromal-BCP-ALL co-cultures inhibited BCP-ALL migration and adhesion. Moreover, these compounds induced anti-leukemic responses in BCP-ALL cells, including a dose-dependent reduction of viability and proliferation, the induction of apoptosis and, importantly, the inhibition of drug resistance. Collectively, these findings indicate galectin-3 regulates BCP-ALL cell responses to chemotherapy through the interactions between leukemia cells and the stroma, and show that a combination of galectin-3 inhibition with conventional drugs can sensitize the leukemia cells to chemotherapy.
Keywords: B-cell precursor ALL; adhesion; carbohydrate-based galectin-3 inhibitor; drug resistance; galectin; galectin-3; glycomimetic; lgals3; microenvironment; migration; monosaccharide; taloside.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
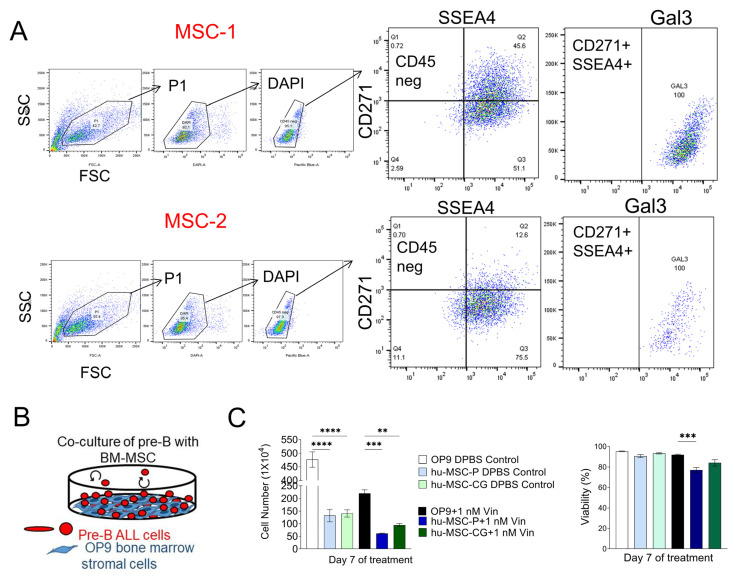







Similar articles
-
Galectin-1 and Galectin-3 in B-Cell Precursor Acute Lymphoblastic Leukemia.Int J Mol Sci. 2022 Nov 18;23(22):14359. doi: 10.3390/ijms232214359. Int J Mol Sci. 2022. PMID: 36430839 Free PMC article.
-
Contribution of the TIME in BCP-ALL: the basis for novel approaches therapeutics.Front Immunol. 2024 Jan 17;14:1325255. doi: 10.3389/fimmu.2023.1325255. eCollection 2023. Front Immunol. 2024. PMID: 38299154 Free PMC article. Review.
-
Treatment of B-cell precursor acute lymphoblastic leukemia with the Galectin-1 inhibitor PTX008.J Exp Clin Cancer Res. 2018 Mar 27;37(1):67. doi: 10.1186/s13046-018-0721-7. J Exp Clin Cancer Res. 2018. PMID: 29580262 Free PMC article.
-
B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3.Oncotarget. 2015 May 10;6(13):11378-94. doi: 10.18632/oncotarget.3409. Oncotarget. 2015. PMID: 25869099 Free PMC article.
-
Galectins as regulators of cell survival in the leukemia niche.Adv Biol Regul. 2019 Jan;71:41-54. doi: 10.1016/j.jbior.2018.09.003. Epub 2018 Sep 12. Adv Biol Regul. 2019. PMID: 30245264 Review.
Cited by
-
Galectin-1 and Galectin-3 in B-Cell Precursor Acute Lymphoblastic Leukemia.Int J Mol Sci. 2022 Nov 18;23(22):14359. doi: 10.3390/ijms232214359. Int J Mol Sci. 2022. PMID: 36430839 Free PMC article.
-
Galectin Family Members: Emerging Novel Targets for Lymphoma Therapy?Front Oncol. 2022 May 23;12:889034. doi: 10.3389/fonc.2022.889034. eCollection 2022. Front Oncol. 2022. PMID: 35677161 Free PMC article. Review.
-
Contribution of the TIME in BCP-ALL: the basis for novel approaches therapeutics.Front Immunol. 2024 Jan 17;14:1325255. doi: 10.3389/fimmu.2023.1325255. eCollection 2023. Front Immunol. 2024. PMID: 38299154 Free PMC article. Review.
-
Bone Marrow Microenvironment-Induced Chemoprotection in KMT2A Rearranged Pediatric AML Is Overcome by Azacitidine-Panobinostat Combination.Cancers (Basel). 2023 Jun 8;15(12):3112. doi: 10.3390/cancers15123112. Cancers (Basel). 2023. PMID: 37370721 Free PMC article.
-
Characterization of mesenchymal stem cells in pre-B acute lymphoblastic leukemia.Front Cell Dev Biol. 2023 Jan 20;11:1005494. doi: 10.3389/fcell.2023.1005494. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 36743421 Free PMC article.
References
-
- Choi S., Henderson M.J., Kwan E., Beesley A.H., Sutton R., Bahar A.Y., Giles J., Venn N.C., Pozza L.D., Baker D.L., et al. Relapse in children with acute lymphoblastic leukemia involving selection of a preexisting drug-resistant subclone. Blood. 2007;110:632–639. doi: 10.1182/blood-2007-01-067785. - DOI - PubMed
-
- Chiarini F., Lonetti A., Evangelisti C., Buontempo F., Orsini E., Evangelisti C., Cappellini A., Neri L.M., McCubrey J.A., Martelli A.M. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochim. Biophys. Acta. 2016;1863:449–463. doi: 10.1016/j.bbamcr.2015.08.015. - DOI - PubMed
-
- Stamatopoulos B., Meuleman N., De Bruyn C., Pieters K., Mineur P., Le Roy C., Saint-Georges S., Varin-Blank N., Cymbalista F., Bron D., et al. AMD3100 disrupts the cross-talk between chronic lymphocytic leukemia cells and a mesenchymal stromal or nurse-like cell-based microenvironment: Pre-clinical evidence for its association with chronic lymphocytic leukemia treatments. Haematologica. 2012;97:608–615. doi: 10.3324/haematol.2011.052779. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
