

Misfolded G Protein-Coupled Receptors and Endocrine Disease. Molecular Mechanisms and Therapeutic Prospects
- PMID: 34830210
- PMCID: PMC8622668
- DOI: 10.3390/ijms222212329
Misfolded G Protein-Coupled Receptors and Endocrine Disease. Molecular Mechanisms and Therapeutic Prospects
Abstract
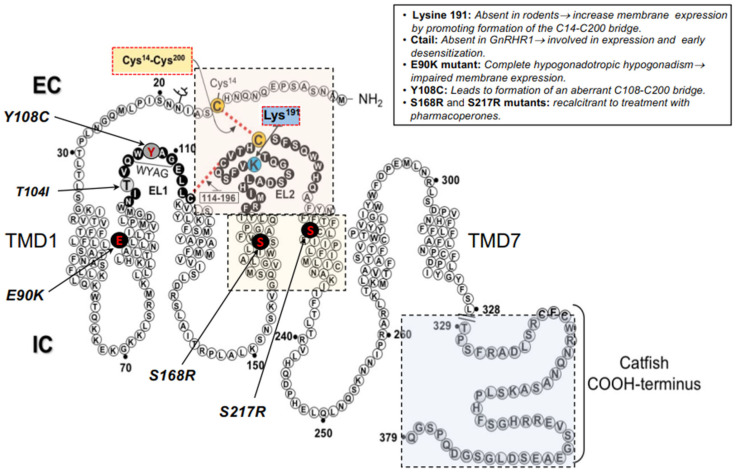
Misfolding of G protein-coupled receptors (GPCRs) caused by mutations frequently leads to disease due to intracellular trapping of the conformationally abnormal receptor. Several endocrine diseases due to inactivating mutations in GPCRs have been described, including X-linked nephrogenic diabetes insipidus, thyroid disorders, familial hypocalciuric hypercalcemia, obesity, familial glucocorticoid deficiency [melanocortin-2 receptor, MC2R (also known as adrenocorticotropin receptor, ACTHR), and reproductive disorders. In these mutant receptors, misfolding leads to endoplasmic reticulum retention, increased intracellular degradation, and deficient trafficking of the abnormal receptor to the cell surface plasma membrane, causing inability of the receptor to interact with agonists and trigger intracellular signaling. In this review, we discuss the mechanisms whereby mutations in GPCRs involved in endocrine function in humans lead to misfolding, decreased plasma membrane expression of the receptor protein, and loss-of-function diseases, and also describe several experimental approaches employed to rescue trafficking and function of the misfolded receptors. Special attention is given to misfolded GPCRs that regulate reproductive function, given the key role played by these particular membrane receptors in sexual development and fertility, and recent reports on promising therapeutic interventions targeting trafficking of these defective proteins to rescue completely or partially their normal function.
Keywords: G protein-coupled receptors; GPCR; GnRHR; gonadotropin receptors; gonadotropin-releasing hormone receptor; loss-of-function diseases; mutations in GPCRs; protein misfolding.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




Similar articles
-
Targeting trafficking as a therapeutic avenue for misfolded GPCRs leading to endocrine diseases.Front Endocrinol (Lausanne). 2022 Aug 25;13:934685. doi: 10.3389/fendo.2022.934685. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36093106 Free PMC article. Review.
-
Pharmacological chaperones correct misfolded GPCRs and rescue function: protein trafficking as a therapeutic target.Subcell Biochem. 2012;63:263-89. doi: 10.1007/978-94-007-4765-4_14. Subcell Biochem. 2012. PMID: 23161143 Review.
-
Mutations in G protein-coupled receptors that impact receptor trafficking and reproductive function.Mol Cell Endocrinol. 2014 Jan 25;382(1):411-423. doi: 10.1016/j.mce.2013.06.024. Epub 2013 Jun 24. Mol Cell Endocrinol. 2014. PMID: 23806559 Free PMC article. Review.
-
Pharmacological chaperones for misfolded gonadotropin-releasing hormone receptors.Adv Pharmacol. 2011;62:109-41. doi: 10.1016/B978-0-12-385952-5.00008-7. Adv Pharmacol. 2011. PMID: 21907908 Free PMC article. Review.
-
Intracellular Trafficking of Gonadotropin Receptors in Health and Disease.Handb Exp Pharmacol. 2018;245:1-39. doi: 10.1007/164_2017_49. Handb Exp Pharmacol. 2018. PMID: 29063275 Review.
Cited by
-
Genetic variants of G-protein coupled receptors associated with pubertal disorders.Reprod Med Biol. 2023 Apr 27;22(1):e12515. doi: 10.1002/rmb2.12515. eCollection 2023 Jan-Dec. Reprod Med Biol. 2023. PMID: 37122876 Free PMC article. Review.
-
Deciphering the 3D Structural Characterization of Gonadotropin-Releasing Hormone in Tenualosa ilisha Using Homology Modeling, Molecular Dynamics, and Docking Approaches.Int J Mol Sci. 2025 Jun 25;26(13):6098. doi: 10.3390/ijms26136098. Int J Mol Sci. 2025. PMID: 40649887 Free PMC article.
-
Human C1orf27 protein interacts with α2A-adrenergic receptor and regulates its anterograde transport.J Biol Chem. 2022 Jun;298(6):102021. doi: 10.1016/j.jbc.2022.102021. Epub 2022 May 10. J Biol Chem. 2022. PMID: 35551911 Free PMC article.
-
Allosteric Regulation of G-Protein-Coupled Receptors: From Diversity of Molecular Mechanisms to Multiple Allosteric Sites and Their Ligands.Int J Mol Sci. 2023 Mar 24;24(7):6187. doi: 10.3390/ijms24076187. Int J Mol Sci. 2023. PMID: 37047169 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
