

The Somatic Mutation Paradigm in Congenital Malformations: Hirschsprung Disease as a Model
- PMID: 34830235
- PMCID: PMC8624421
- DOI: 10.3390/ijms222212354
The Somatic Mutation Paradigm in Congenital Malformations: Hirschsprung Disease as a Model
Abstract
Patients with Hirschsprung disease (HSCR) do not always receive a genetic diagnosis after routine screening in clinical practice. One of the reasons for this could be that the causal mutation is not present in the cell types that are usually tested-whole blood, dermal fibroblasts or saliva-but is only in the affected tissue. Such mutations are called somatic, and can occur in a given cell at any stage of development after conception. They will then be present in all subsequent daughter cells. Here, we investigated the presence of somatic mutations in HSCR patients. For this, whole-exome sequencing and copy number analysis were performed in DNA isolated from purified enteric neural crest cells (ENCCs) and blood or fibroblasts of the same patient. Variants identified were subsequently validated by Sanger sequencing. Several somatic variants were identified in all patients, but causative mutations for HSCR were not specifically identified in the ENCCs of these patients. Larger copy number variants were also not found to be specific to ENCCs. Therefore, we believe that somatic mutations are unlikely to be identified, if causative for HSCR. Here, we postulate various modes of development following the occurrence of a somatic mutation, to describe the challenges in detecting such mutations, and hypothesize how somatic mutations may contribute to 'missing heritability' in developmental defects.
Keywords: Enteric Nervous System; Hirschsprung disease; developmental defects; gastrointestinal disease; missing heritability; motility disorder; somatic mutation.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the study design, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Figures
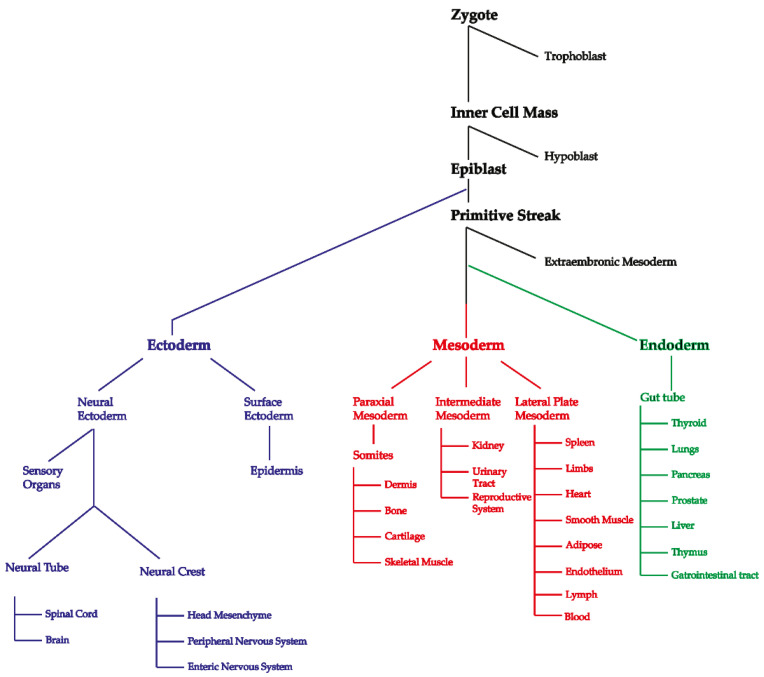
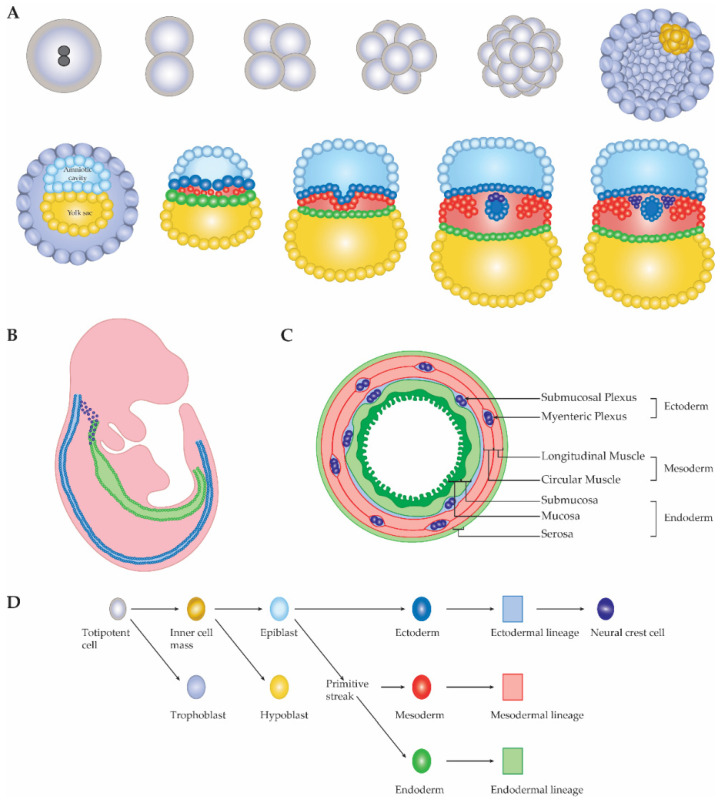
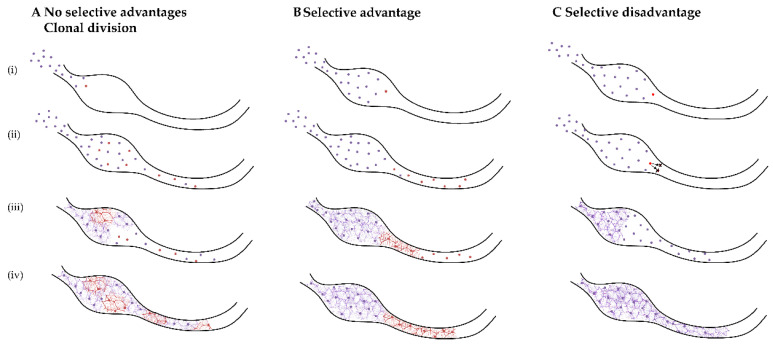
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
