

Impact of Chromatin Dynamics and DNA Repair on Genomic Stability and Treatment Resistance in Pediatric High-Grade Gliomas
- PMID: 34830833
- PMCID: PMC8616465
- DOI: 10.3390/cancers13225678
Impact of Chromatin Dynamics and DNA Repair on Genomic Stability and Treatment Resistance in Pediatric High-Grade Gliomas
Abstract
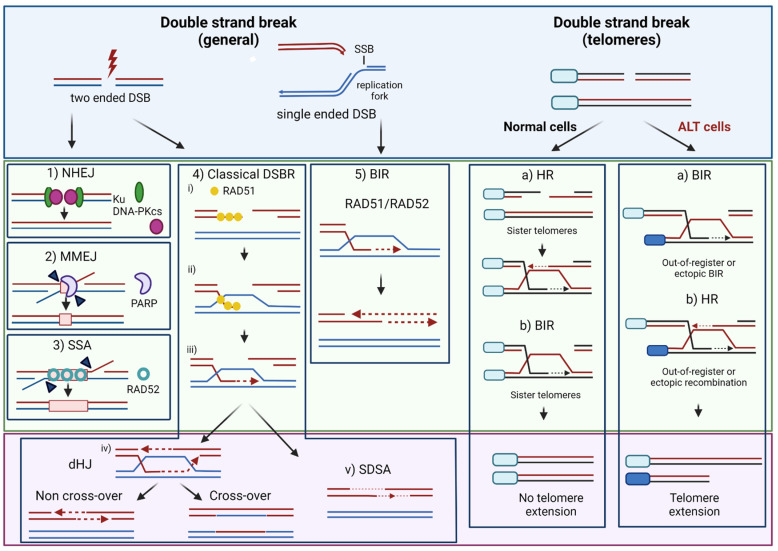
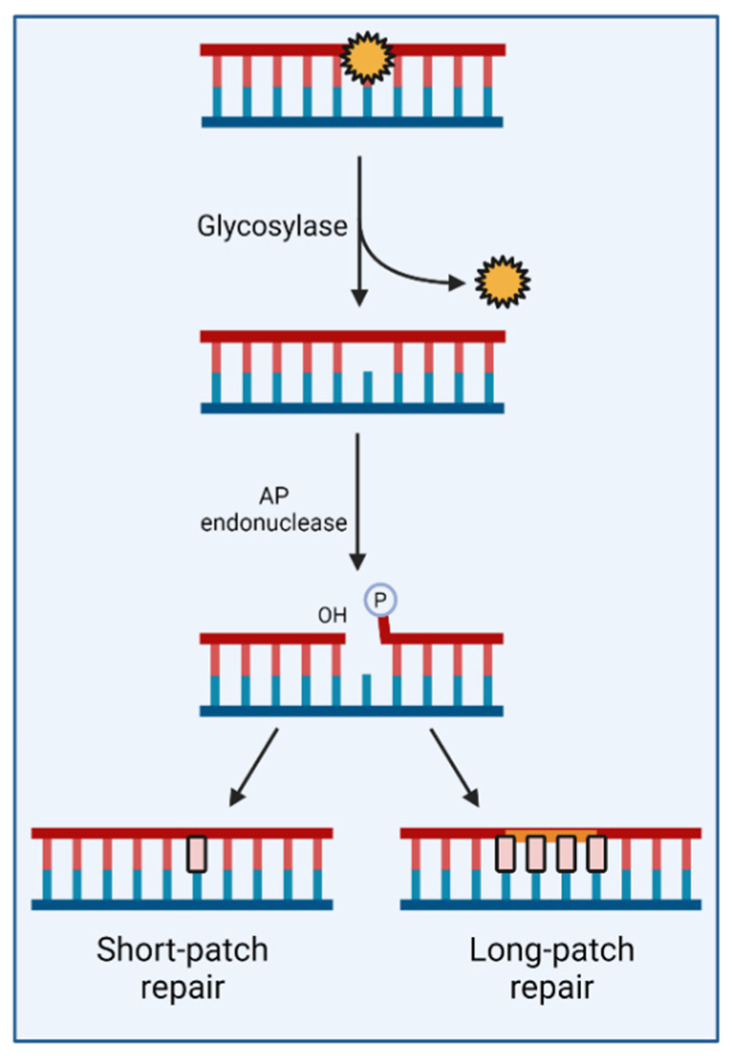
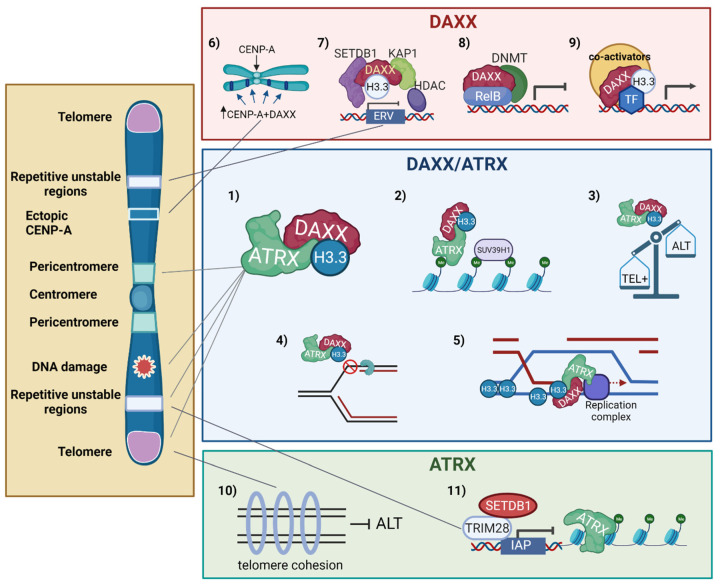
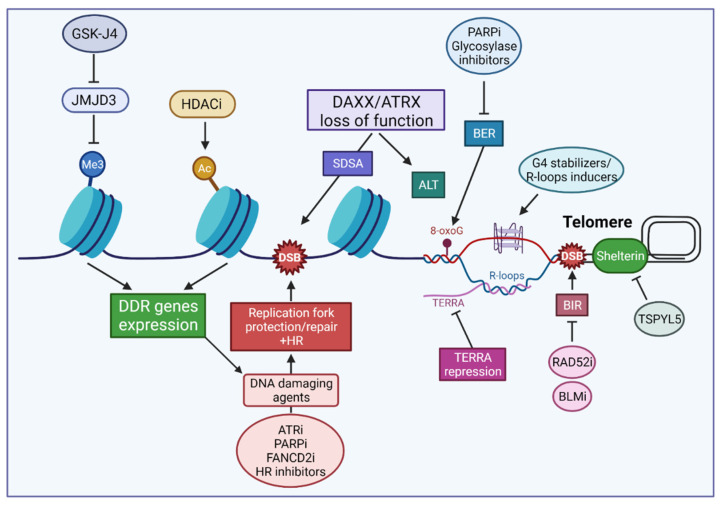
Despite their low incidence, pediatric high-grade gliomas (pHGGs), including diffuse intrinsic pontine gliomas (DIPGs), are the leading cause of mortality in pediatric neuro-oncology. Recurrent, mutually exclusive mutations affecting K27 (K27M) and G34 (G34R/V) in the N-terminal tail of histones H3.3 and H3.1 act as key biological drivers of pHGGs. Notably, mutations in H3.3 are frequently associated with mutations affecting ATRX and DAXX, which encode a chaperone complex that deposits H3.3 into heterochromatic regions, including telomeres. The K27M and G34R/V mutations lead to distinct epigenetic reprogramming, telomere maintenance mechanisms, and oncogenesis scenarios, resulting in distinct subgroups of patients characterized by differences in tumor localization, clinical outcome, as well as concurrent epigenetic and genetic alterations. Contrasting with our understanding of the molecular biology of pHGGs, there has been little improvement in the treatment of pHGGs, with the current mainstays of therapy-genotoxic chemotherapy and ionizing radiation (IR)-facing the development of tumor resistance driven by complex DNA repair pathways. Chromatin and nucleosome dynamics constitute important modulators of the DNA damage response (DDR). Here, we summarize the major DNA repair pathways that contribute to resistance to current DNA damaging agent-based therapeutic strategies and describe the telomere maintenance mechanisms encountered in pHGGs. We then review the functions of H3.3 and its chaperones in chromatin dynamics and DNA repair, as well as examining the impact of their mutation/alteration on these processes. Finally, we discuss potential strategies targeting DNA repair and epigenetic mechanisms as well as telomere maintenance mechanisms, to improve the treatment of pHGGs.
Keywords: (peri)centromere; ATRX; DAXX; DNA repair; alternative lengthening of telomere (ALT); chemoresistance; chromatin dynamics; genomic instability; pediatric high-grade gliomas; synthetic lethality; telomerase; telomere; variant H3.3 histone.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hoffman L.M., Veldhuijzen van Zanten S.E.M., Colditz N., Baugh J., Chaney B., Hoffmann M., Lane A., Fuller C., Miles L., Hawkins C., et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018;36:1963–1972. doi: 10.1200/JCO.2017.75.9308. - DOI - PMC - PubMed
-
- Mackay A., Burford A., Carvalho D., Izquierdo E., Fazal-Salom J., Taylor K.R., Bjerke L., Clarke M., Vinci M., Nandhabalan M., et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell. 2017;32:520–537.e5. doi: 10.1016/j.ccell.2017.08.017. - DOI - PMC - PubMed
-
- Sturm D., Witt H., Hovestadt V., Khuong-Quang D.A., Jones D.T., Konermann C., Pfaff E., Tonjes M., Sill M., Bender S., et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
