

CFTR Protein: Not Just a Chloride Channel?
- PMID: 34831067
- PMCID: PMC8616376
- DOI: 10.3390/cells10112844
CFTR Protein: Not Just a Chloride Channel?
Abstract
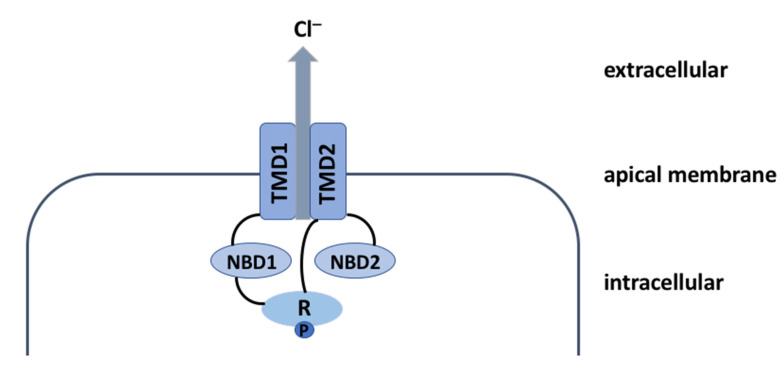
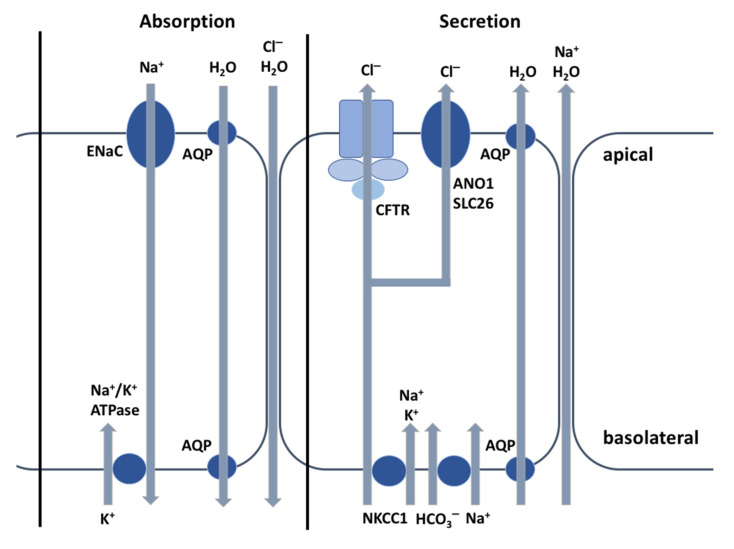
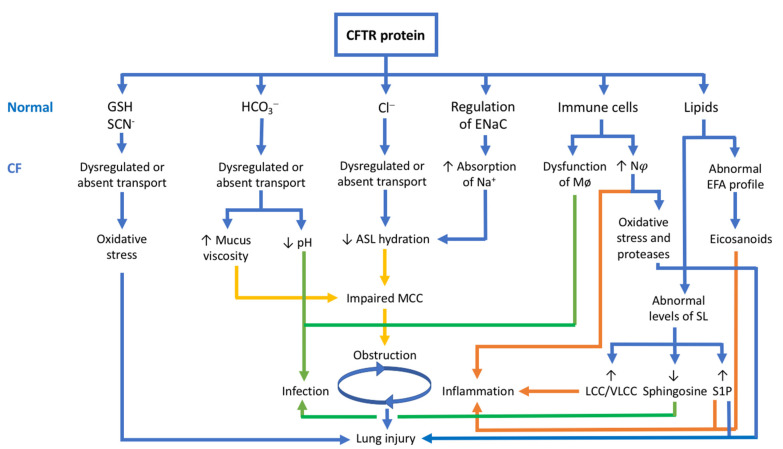
Cystic fibrosis (CF) is a recessive genetic disease caused by mutations in a gene encoding a protein called Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). The CFTR protein is known to acts as a chloride (Cl-) channel expressed in the exocrine glands of several body systems where it also regulates other ion channels, including the epithelial sodium (Na+) channel (ENaC) that plays a key role in salt absorption. This function is crucial to the osmotic balance of the mucus and its viscosity. However, the pathophysiology of CF is more challenging than a mere dysregulation of epithelial ion transport, mainly resulting in impaired mucociliary clearance (MCC) with consecutive bronchiectasis and in exocrine pancreatic insufficiency. This review shows that the CFTR protein is not just a chloride channel. For a long time, research in CF has focused on abnormal Cl- and Na+ transport. Yet, the CFTR protein also regulates numerous other pathways, such as the transport of HCO3-, glutathione and thiocyanate, immune cells, and the metabolism of lipids. It influences the pH homeostasis of airway surface liquid and thus the MCC as well as innate immunity leading to chronic infection and inflammation, all of which are considered as key pathophysiological characteristics of CF.
Keywords: CFTR protein; bicarbonate; channel; chloride; cystic fibrosis; glutathione; lipids; macrophages; neutrophils; thiocyanate.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Andersen D.H. Cystic fibrosis of the pancreas and its relation to celiac diseasea clinical and pathologic study. Am. J. Dis. Child. 1938;56:344–399. doi: 10.1001/archpedi.1938.01980140114013. - DOI
-
- Fanconi G., Uehlinger E., Knauer C. Das Coeliakie-syndrom bei angeborener zystischer Pankreasfibromatose und Bronchiektasien. Wien. Med. Wchnschr. 1936;86:753–756.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
