

Cortactin Modulates Lung Endothelial Apoptosis Induced by Cigarette Smoke
- PMID: 34831092
- PMCID: PMC8616125
- DOI: 10.3390/cells10112869
Cortactin Modulates Lung Endothelial Apoptosis Induced by Cigarette Smoke
Abstract
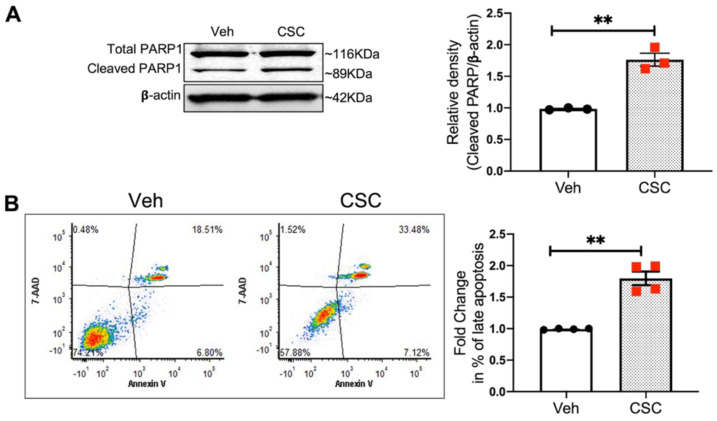
Cigarette smoke (CS) is the primary cause of Chronic Obstructive Pulmonary Disease (COPD), and an important pathophysiologic event in COPD is CS-induced apoptosis in lung endothelial cells (EC). Cortactin (CTTN) is a cytoskeletal actin-binding regulatory protein with modulation by Src-mediated tyrosine phosphorylation. Based upon data demonstrating reduced CTTN mRNA levels in the lungs of smokers compared to non-smokers, we hypothesized a functional role for CTTN in CS-induced mitochondrial ROS generation and apoptosis in lung EC. Exposure of cultured human lung EC to CS condensate (CSC) led to the rearrangement of the actin cytoskeleton and increased CTTN tyrosine phosphorylation (within hours). Exposure to CS significantly increased EC mitochondrial ROS generation and EC apoptosis. The functional role of CTTN in these CSC-induced EC responses was explored using cortactin siRNA to reduce its expression, and by using a blocking peptide for the CTTN SH3 domain, which is critical to cytoskeletal interactions. CTTN siRNA or blockade of its SH3 domain resulted in significantly increased EC mitochondrial ROS and apoptosis and augmented CSC-induced effects. Exposure of lung EC to e-cigarette condensate demonstrated similar results, with CTTN siRNA or SH3 domain blocking peptide increasing lung EC apoptosis. These data demonstrate a novel role for CTTN in modulating lung EC apoptosis induced by CS or e-cigarettes potentially providing new insights into COPD pathogenesis.
Keywords: COPD; cytoskeleton; e-cigarette; endothelium; lung injury; mitochondrial ROS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures









References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
