

Mitochondria in Diabetic Kidney Disease
- PMID: 34831168
- PMCID: PMC8616075
- DOI: 10.3390/cells10112945
Mitochondria in Diabetic Kidney Disease
Abstract
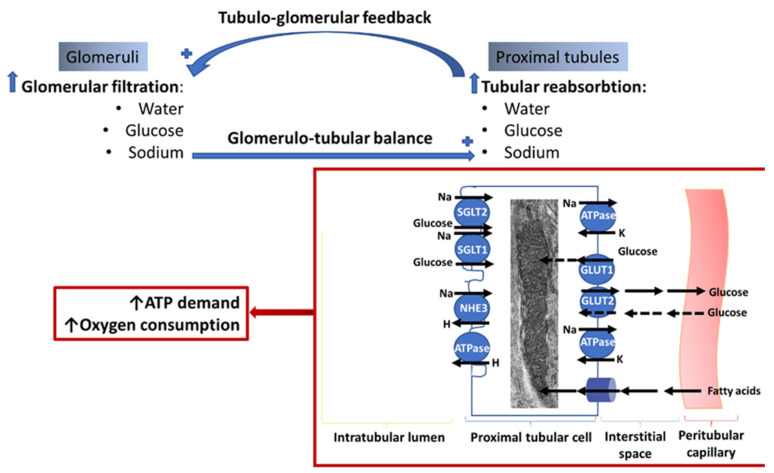
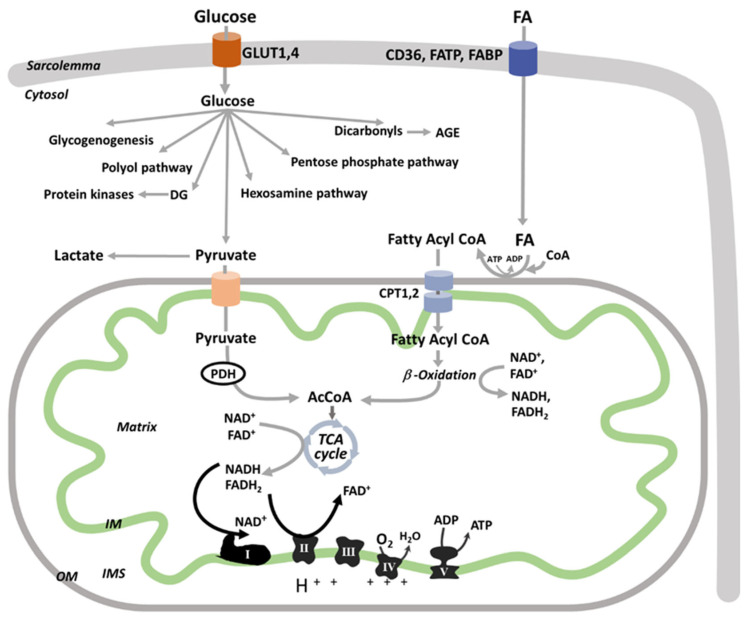
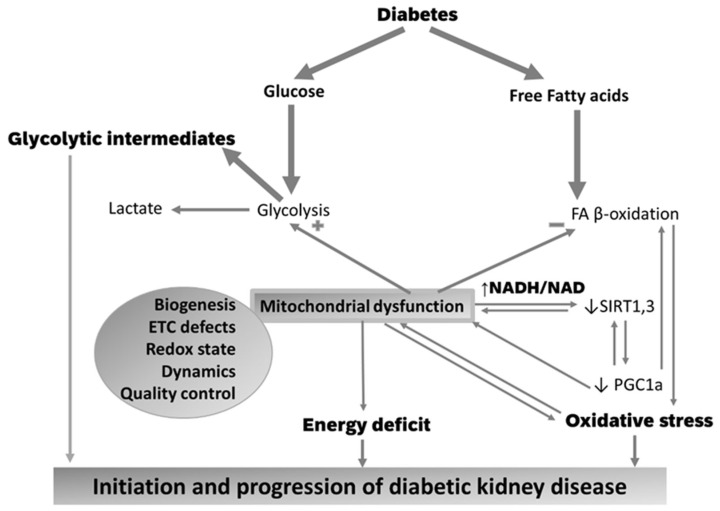
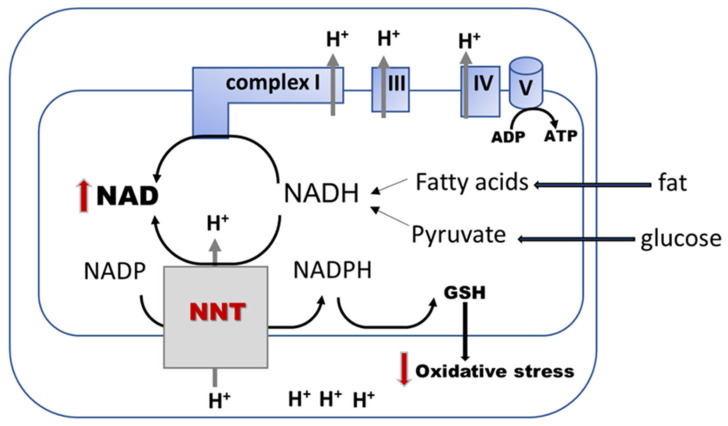
Diabetic kidney disease (DKD) is the leading cause of end stage renal disease (ESRD) in the USA. The pathogenesis of DKD is multifactorial and involves activation of multiple signaling pathways with merging outcomes including thickening of the basement membrane, podocyte loss, mesangial expansion, tubular atrophy, and interstitial inflammation and fibrosis. The glomerulo-tubular balance and tubule-glomerular feedback support an increased glomerular filtration and tubular reabsorption, with the latter relying heavily on ATP and increasing the energy demand. There is evidence that alterations in mitochondrial bioenergetics in kidney cells lead to these pathologic changes and contribute to the progression of DKD towards ESRD. This review will focus on the dialogue between alterations in bioenergetics in glomerular and tubular cells and its role in the development of DKD. Alterations in energy substrate selection, electron transport chain, ATP generation, oxidative stress, redox status, protein posttranslational modifications, mitochondrial dynamics, and quality control will be discussed. Understanding the role of bioenergetics in the progression of diabetic DKD may provide novel therapeutic approaches to delay its progression to ESRD.
Keywords: bioenergetics; diabetes; diabetic kidney disease; mitochondria; redox status.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Pugliese G., Penno G., Natali A., Barutta F., Di Paolo S., Reboldi G., Gesualdo L., De Nicola L., Italian Diabetes Society. the Italian Society of Nephrology Diabetic kidney disease: New clinical and therapeutic issues. Joint position statement of the Italian Diabetes Society and the Italian Society of Nephrology on “The natural history of diabetic kidney disease and treatment of hyperglycemia in patients with type 2 diabetes and impaired renal function”. Nutr. Metab. Cardiovasc. Dis. 2019;29:1127–1150. doi: 10.1016/j.numecd.2019.07.017. - DOI - PubMed
-
- Minutolo R., Gabbai F.B., Provenzano M., Chiodini P., Borrelli S., Garofalo C., Sasso F.C., Santoro D., Bellizzi V., Conte G., et al. Cardiorenal prognosis by residual proteinuria level in diabetic chronic kidney disease: Pooled analysis of four cohort studies. Nephrol. Dial. Transplant. 2018;33:1942–1949. doi: 10.1093/ndt/gfy032. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
