

TRPV4 Mechanotransduction in Fibrosis
- PMID: 34831281
- PMCID: PMC8619244
- DOI: 10.3390/cells10113053
TRPV4 Mechanotransduction in Fibrosis
Abstract
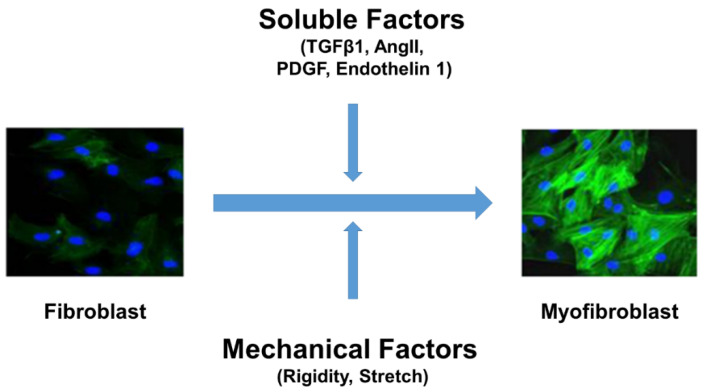
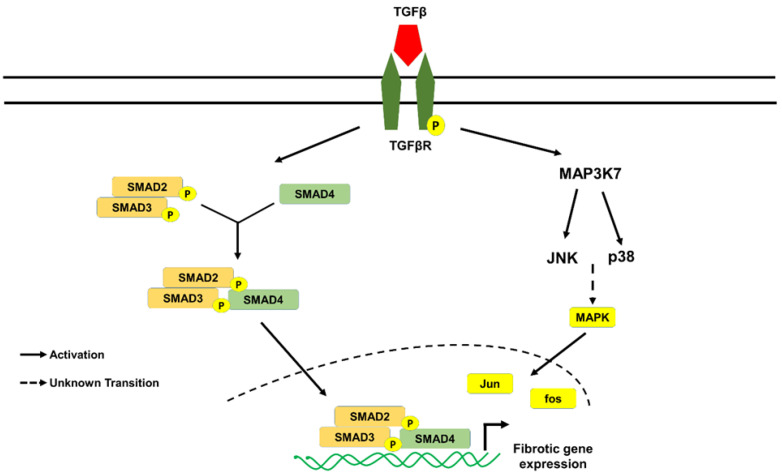
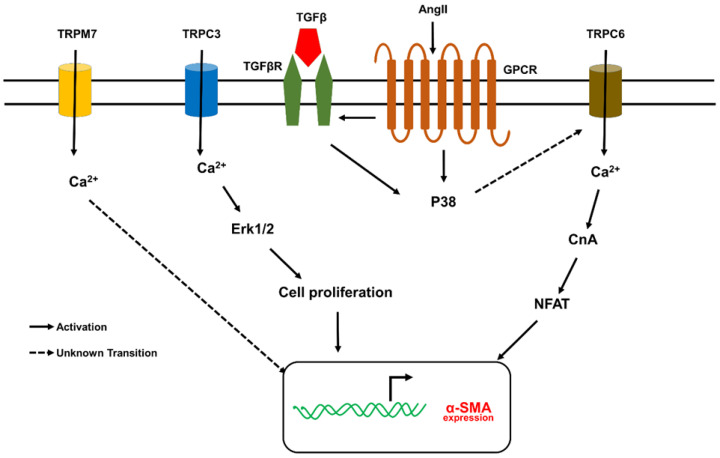
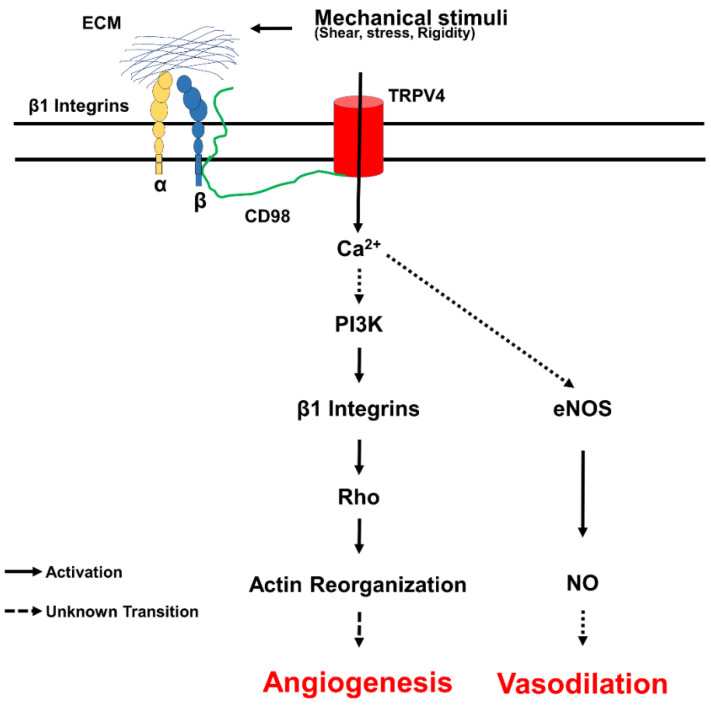
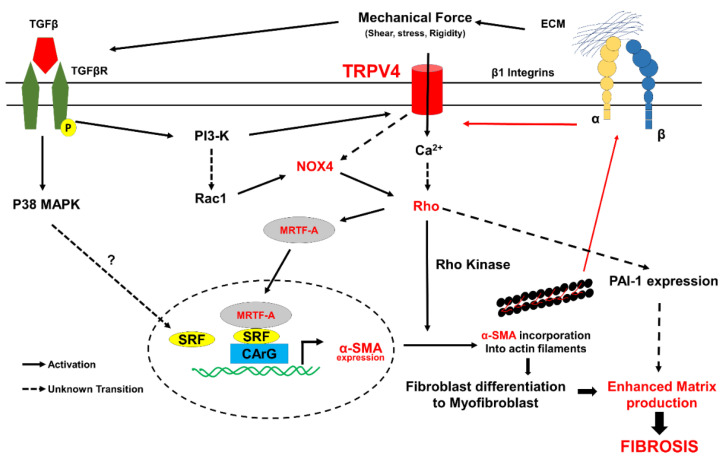
Fibrosis is an irreversible, debilitating condition marked by the excessive production of extracellular matrix and tissue scarring that eventually results in organ failure and disease. Differentiation of fibroblasts to hypersecretory myofibroblasts is the key event in fibrosis. Although both soluble and mechanical factors are implicated in fibroblast differentiation, much of the focus is on TGF-β signaling, but to date, there are no specific drugs available for the treatment of fibrosis. In this review, we describe the role for TRPV4 mechanotransduction in cardiac and lung fibrosis, and we propose TRPV4 as an alternative therapeutic target for fibrosis.
Keywords: TGF-β; TRPV4; calcium; extracellular matrix; fibroblast; fibrosis; mechanotransduction; myofibroblast.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
