

Transcriptional Profiles of Cell Fate Transitions Reveal Early Drivers of Neuronal Apoptosis and Survival
- PMID: 34831459
- PMCID: PMC8620386
- DOI: 10.3390/cells10113238
Transcriptional Profiles of Cell Fate Transitions Reveal Early Drivers of Neuronal Apoptosis and Survival
Abstract
Neuronal apoptosis and survival are regulated at the transcriptional level. To identify key genes and upstream regulators primarily responsible for these processes, we overlayed the temporal transcriptome of cerebellar granule neurons following induction of apoptosis and their rescue by three different neurotrophic factors. We identified a core set of 175 genes showing opposite expression trends at the intersection of apoptosis and survival. Their functional annotations and expression signatures significantly correlated to neurological, psychiatric and oncological disorders. Transcription regulatory network analysis revealed the action of nine upstream transcription factors, converging pro-apoptosis and pro-survival-inducing signals in a highly interconnected functionally and temporally ordered manner. Five of these transcription factors are potential drug targets. Transcriptome-based computational drug repurposing produced a list of drug candidates that may revert the apoptotic core set signature. Besides elucidating early drivers of neuronal apoptosis and survival, our systems biology-based perspective paves the way to innovative pharmacology focused on upstream targets and regulatory networks.
Keywords: apoptosis; disease; drug repurposing; drug targets; functional enrichment; neurotrophic factors; regulatory network; survival; transcriptional analysis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
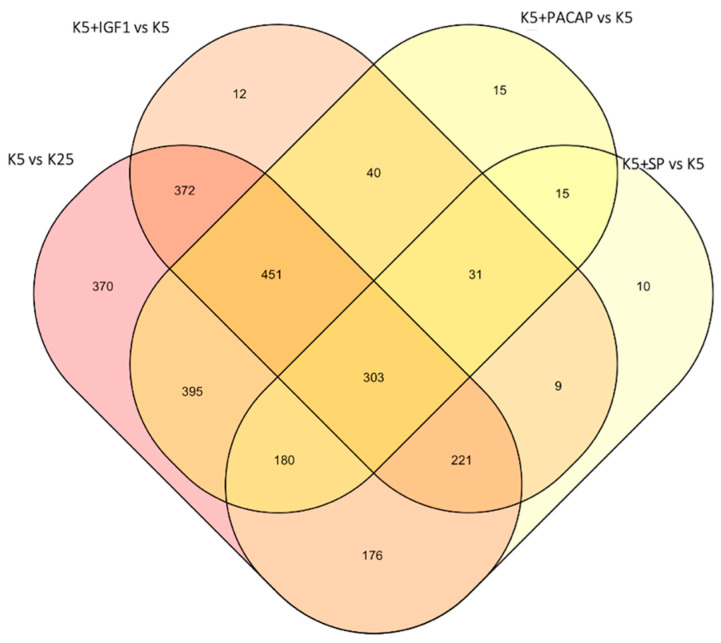
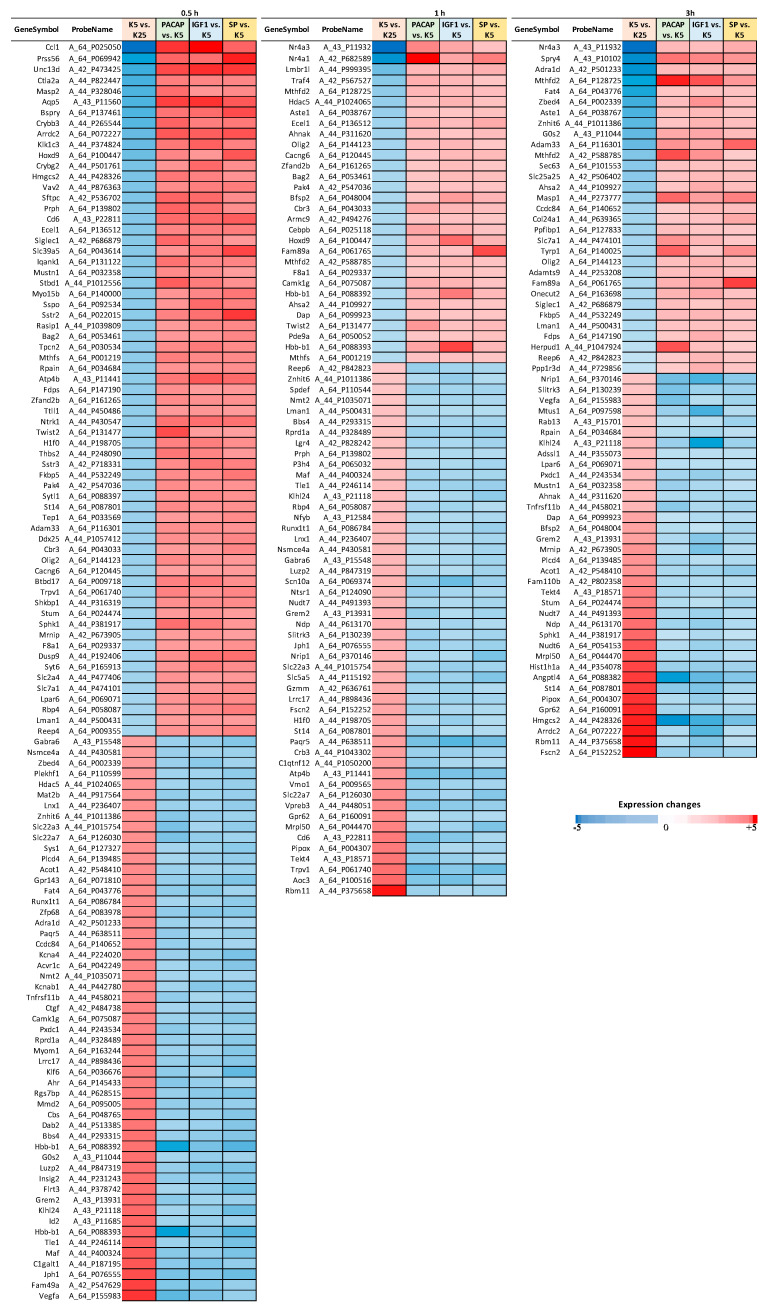
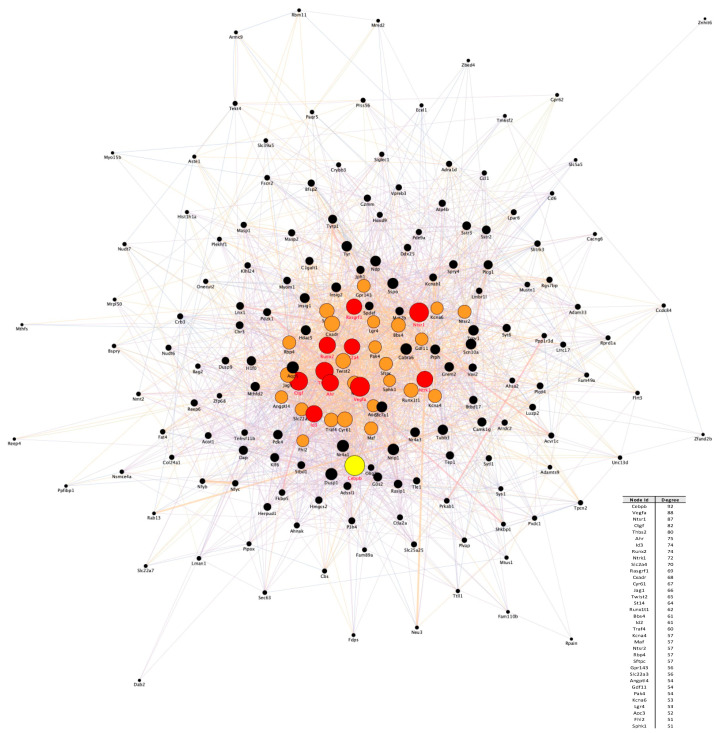
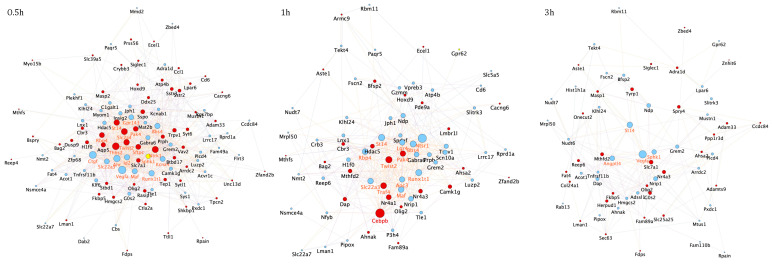
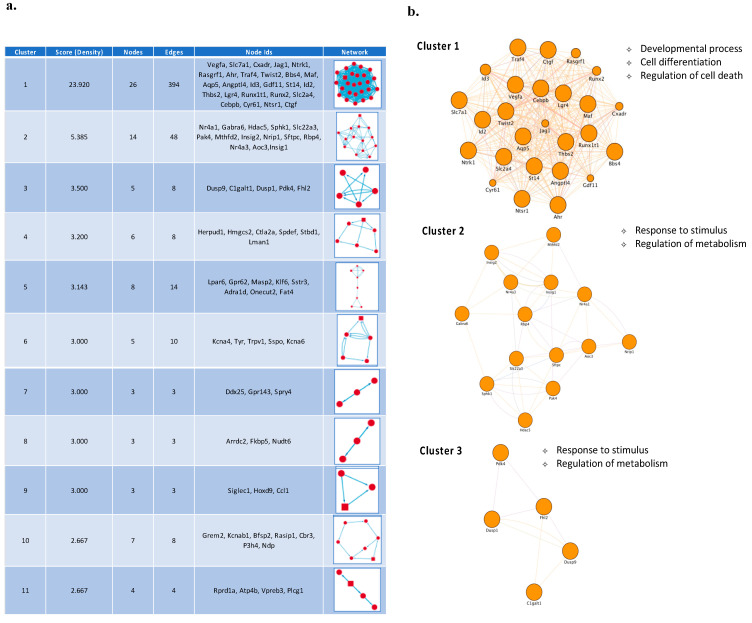
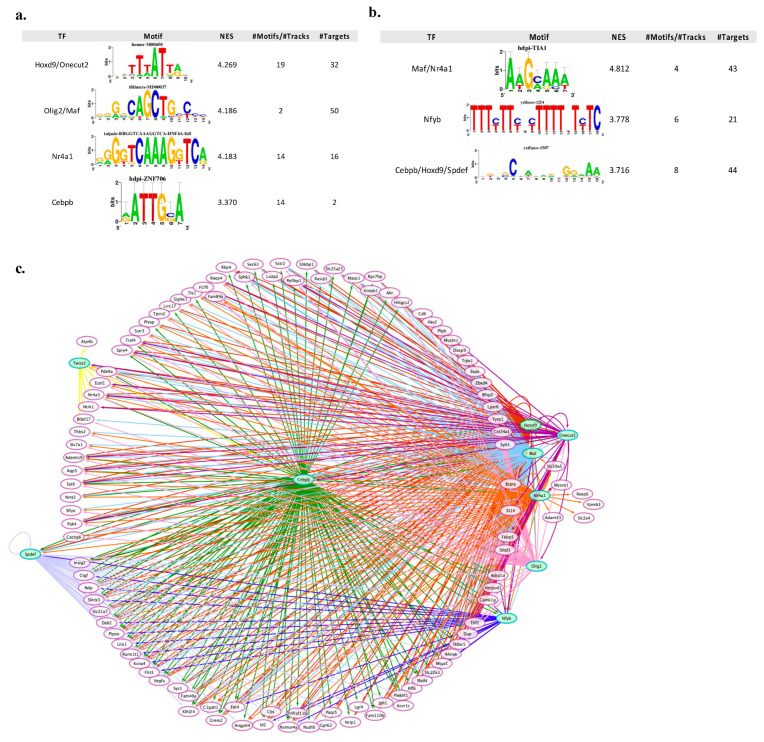
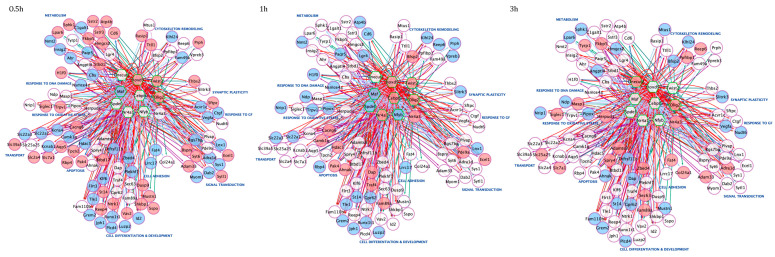
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
