

Genomic Analysis of Prophages from Klebsiella pneumoniae Clinical Isolates
- PMID: 34835377
- PMCID: PMC8617712
- DOI: 10.3390/microorganisms9112252
Genomic Analysis of Prophages from Klebsiella pneumoniae Clinical Isolates
Abstract
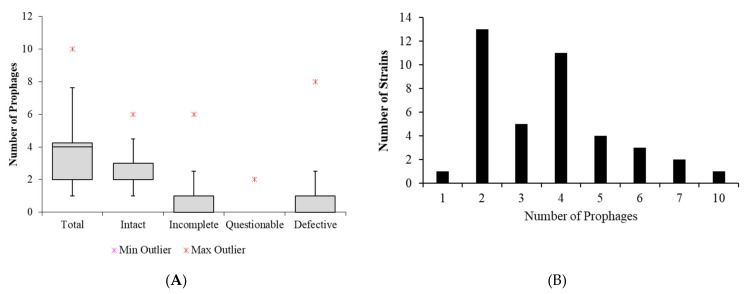
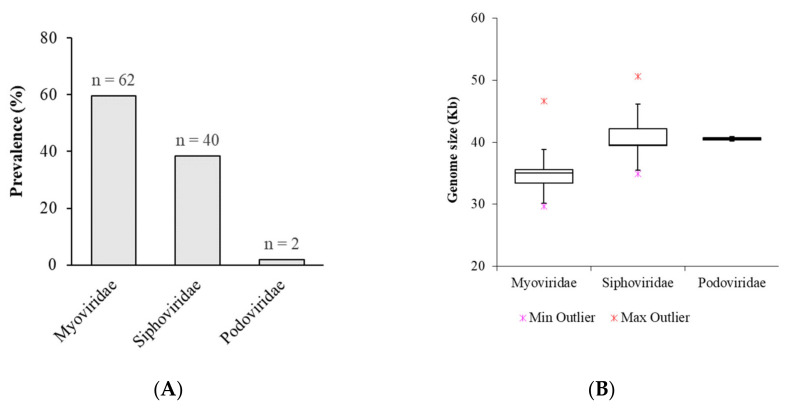
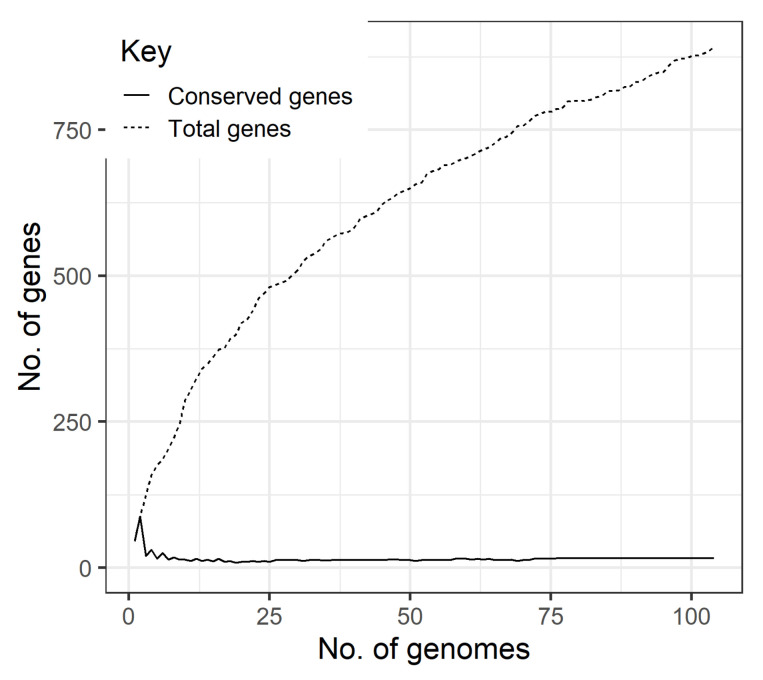
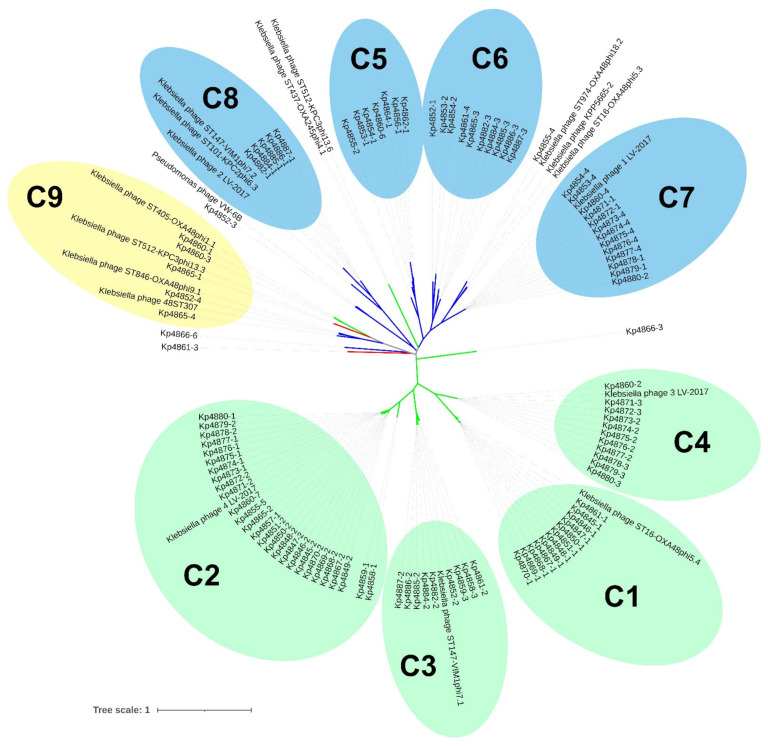
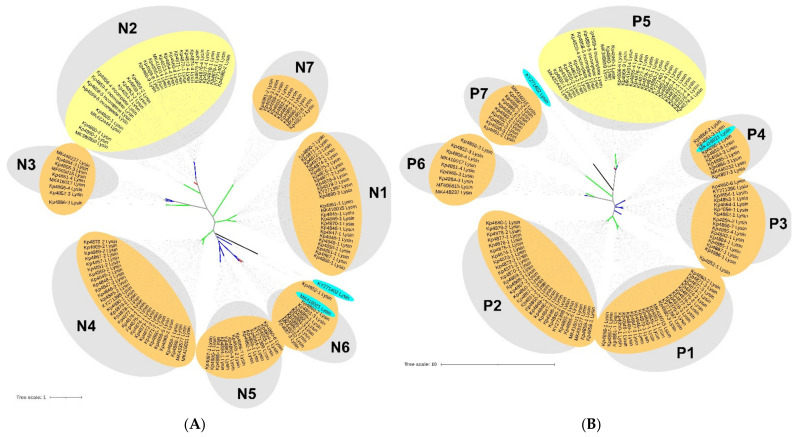
Klebsiella pneumoniae is an increasing threat to public health and represents one of the most concerning pathogens involved in life-threatening infections. The resistant and virulence determinants are coded by mobile genetic elements which can easily spread between bacteria populations and co-evolve with its genomic host. In this study, we present the full genomic sequences, insertion sites and phylogenetic analysis of 150 prophages found in 40 K. pneumoniae clinical isolates obtained from an outbreak in a Portuguese hospital. All strains harbored at least one prophage and we identified 104 intact prophages (69.3%). The prophage size ranges from 29.7 to 50.6 kbp, coding between 32 and 78 putative genes. The prophage GC content is 51.2%, lower than the average GC content of 57.1% in K. pneumoniae. Complete prophages were classified into three families in the order Caudolovirales: Myoviridae (59.6%), Siphoviridae (38.5%) and Podoviridae (1.9%). In addition, an alignment and phylogenetic analysis revealed nine distinct clusters. Evidence of recombination was detected within the genome of some prophages but, in most cases, proteins involved in viral structure, transcription, replication and regulation (lysogenic/lysis) were maintained. These results support the knowledge that prophages are diverse and widely disseminated in K. pneumoniae genomes, contributing to the evolution of this species and conferring additional phenotypes. Moreover, we identified K. pneumoniae prophages in a set of endolysin genes, which were found to code for proteins with lysozyme activity, cleaving the β-1,4 linkages between N-acetylmuramic acid and N-acetyl-D-glucosamine residues in the peptidoglycan network and thus representing genes with the potential for lysin phage therapy.
Keywords: K. pneumoniae genomes; bacteriophage; bioinformatics; comparative genomics; genomic analysis; phage endolysins; phylogeny; prophages; sequence annotation and comparison.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Coovadia Y.M., Johnson A.P., Bhana R.H., Hutchinson G.R., George R.C., Hafferjee I.E. Multiresistant Klebsiella pneumoniae in a neonatal nursery: The importance of maintenance of infection control policies and procedures in the prevention of outbreaks. J. Hosp. Infect. 1992;22:197–205. doi: 10.1016/0195-6701(92)90044-M. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
