

Latency-associated DNA methylation patterns among HIV-1 infected individuals with distinct disease progression courses or antiretroviral virologic response
- PMID: 34837007
- PMCID: PMC8626465
- DOI: 10.1038/s41598-021-02463-0
Latency-associated DNA methylation patterns among HIV-1 infected individuals with distinct disease progression courses or antiretroviral virologic response
Abstract
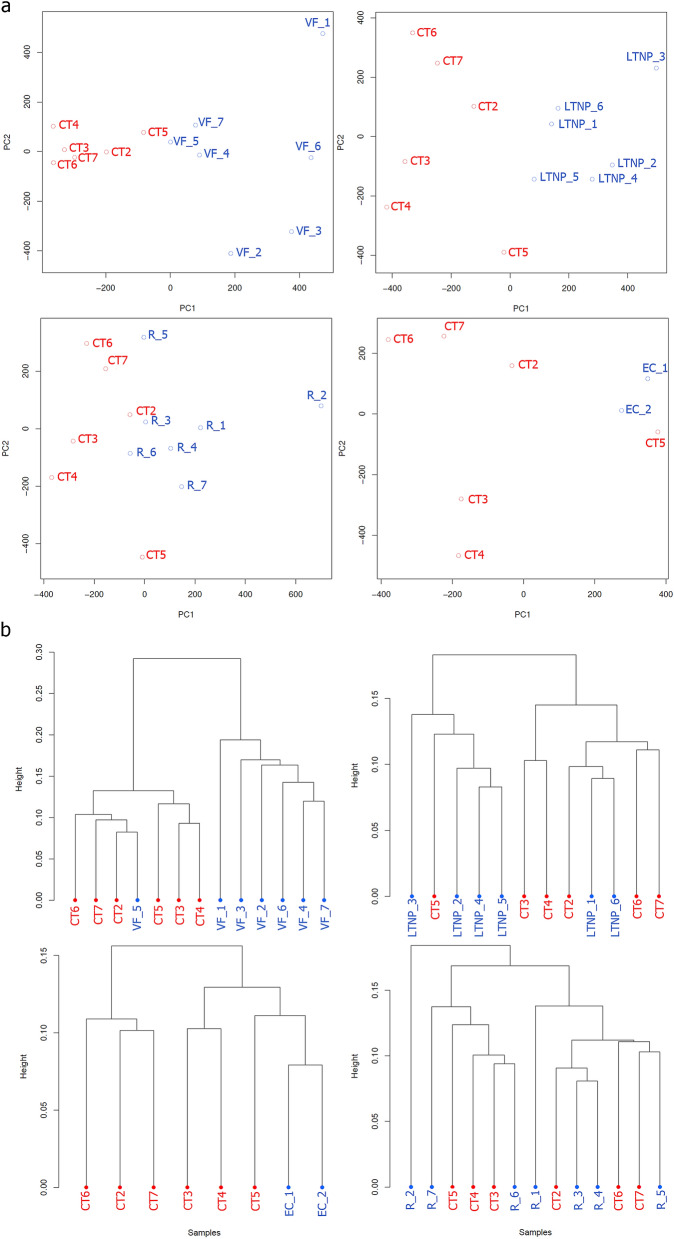
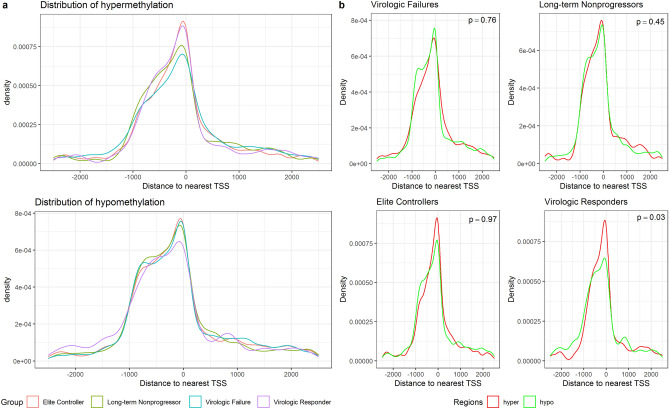
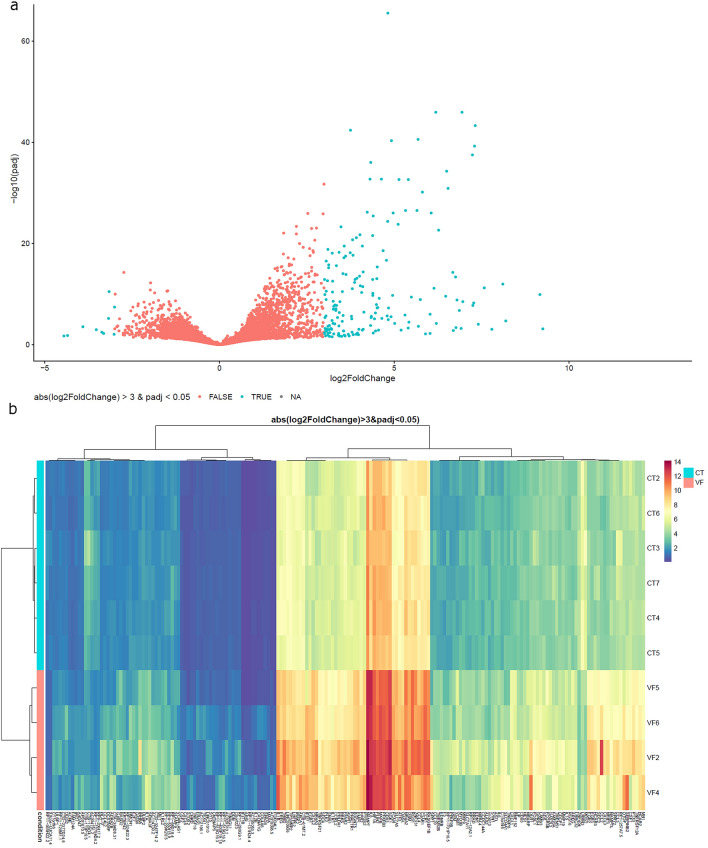
DNA methylation is one of the epigenetic modifications that configures gene transcription programs. This study describes the DNA methylation profile of HIV-infected individuals with distinct characteristics related to natural and artificial viremia control. Sheared DNA from circulating mononuclear cells was subjected to target enrichment bisulfite sequencing designed to cover CpG-rich genomic regions. Gene expression was assessed through RNA-seq. Hypermethylation in virologic responders was highly distributed closer to Transcription Start Sites (p-value = 0.03). Hyper and hypomethylation levels within TSS adjacencies varied according to disease progression status (Kruskal-Wallis, p < 0.001), and specific differentially methylated regions associated genes were identified for each group. The lower the promoter methylation, the higher the gene expression in subjects undergoing virologic failure (R = - 0.82, p = 0.00068). Among the inversely correlated genes, those supporting glycolysis and its related pathways were hypomethylated and up-regulated in virologic failures. Disease progression heterogeneity was associated with distinct DNA methylation patterns in terms of rates and distribution. Methylation was associated with the expression of genes sustaining intracellular glucose metabolism in subjects undergoing antiretroviral virologic failure. Our findings highlight that DNA methylation is associated with latency, disease progression, and fundamental cellular processes.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Suzuki MM, Bird A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
