

Virus-infection in cochlear supporting cells induces audiosensory receptor hair cell death by TRAIL-induced necroptosis
- PMID: 34843580
- PMCID: PMC8629241
- DOI: 10.1371/journal.pone.0260443
Virus-infection in cochlear supporting cells induces audiosensory receptor hair cell death by TRAIL-induced necroptosis
Abstract
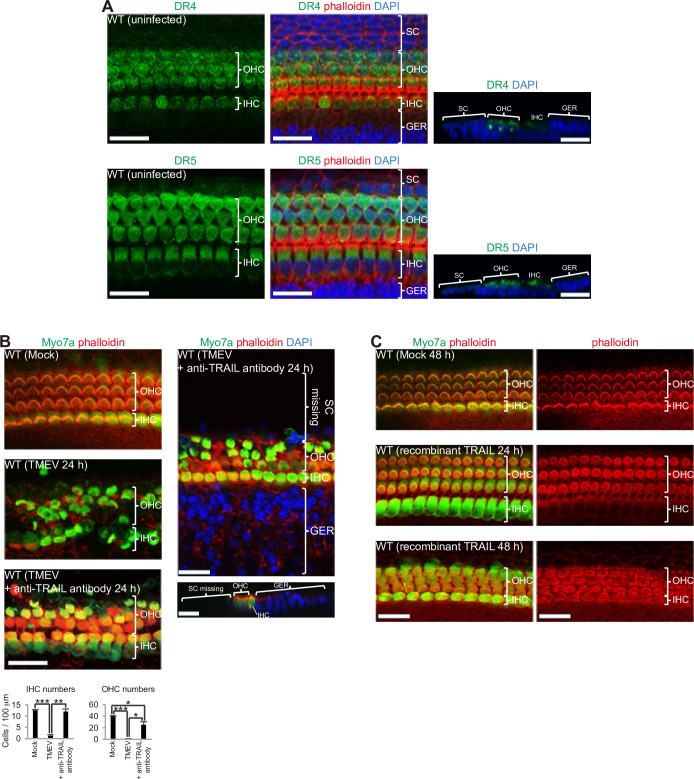
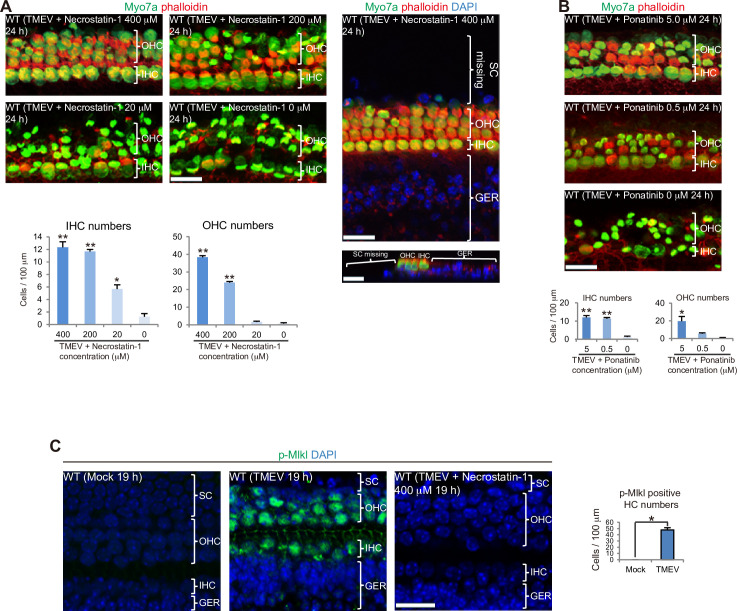
Although sensorineural hearing loss (SHL) is relatively common, its cause has not been identified in most cases. Previous studies have suggested that viral infection is a major cause of SHL, especially sudden SHL, but the system that protects against pathogens in the inner ear, which is isolated by the blood-labyrinthine barrier, remains poorly understood. We recently showed that, as audiosensory receptor cells, cochlear hair cells (HCs) are protected by surrounding accessory supporting cells (SCs) and greater epithelial ridge (GER or Kölliker's organ) cells (GERCs) against viral infections. Here, we found that virus-infected SCs and GERCs induce HC death via production of the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL). Notably, the HCs expressed the TRAIL death receptors (DR) DR4 and DR5, and virus-induced HC death was suppressed by TRAIL-neutralizing antibodies. TRAIL-induced HC death was not caused by apoptosis, and was inhibited by necroptosis inhibitors. Moreover, corticosteroids, the only effective drug for SHL, inhibited the virus-induced transformation of SCs and GERCs into macrophage-like cells and HC death, while macrophage depletion also inhibited virus-induced HC death. These results reveal a novel mechanism underlying virus-induced HC death in the cochlear sensory epithelium and suggest a possible target for preventing virus-induced SHL.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
