

Understanding and targeting prostate cancer cell heterogeneity and plasticity
- PMID: 34844845
- PMCID: PMC9106849
- DOI: 10.1016/j.semcancer.2021.11.001
Understanding and targeting prostate cancer cell heterogeneity and plasticity
Abstract
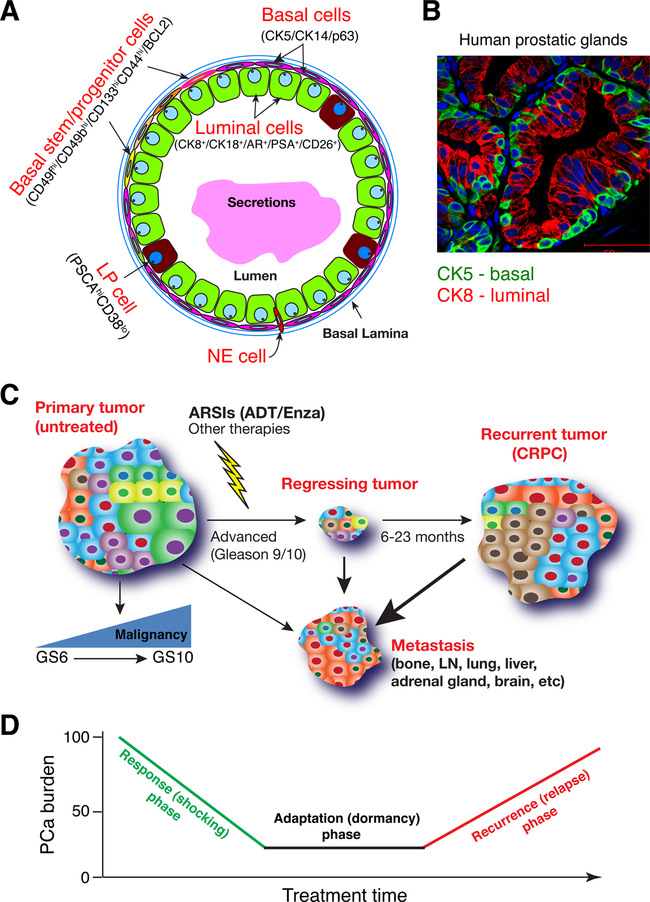
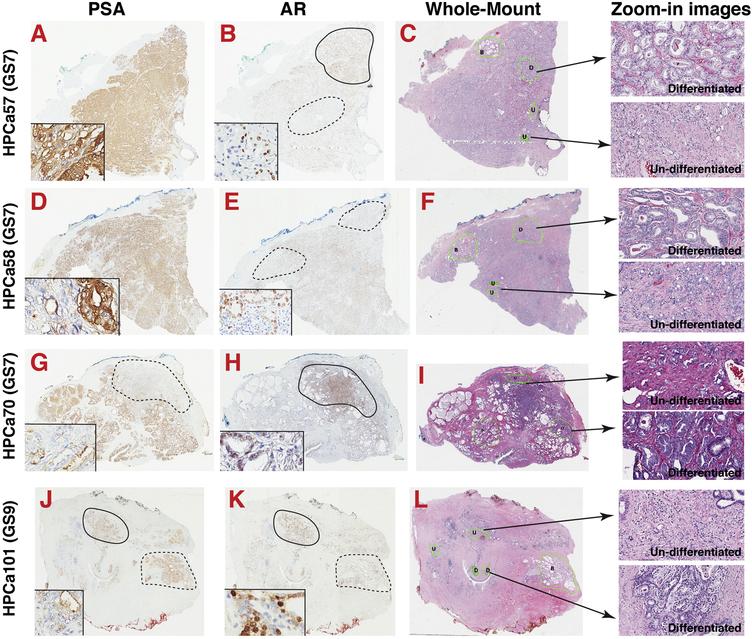
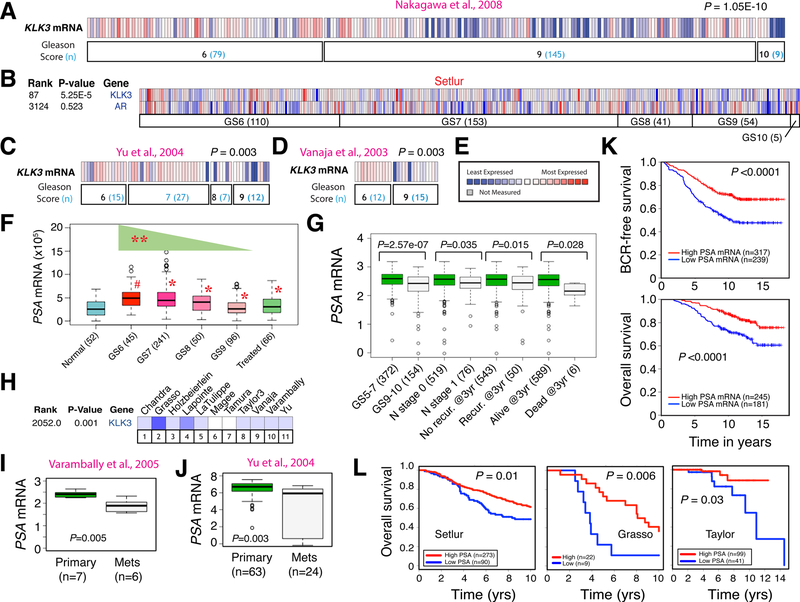
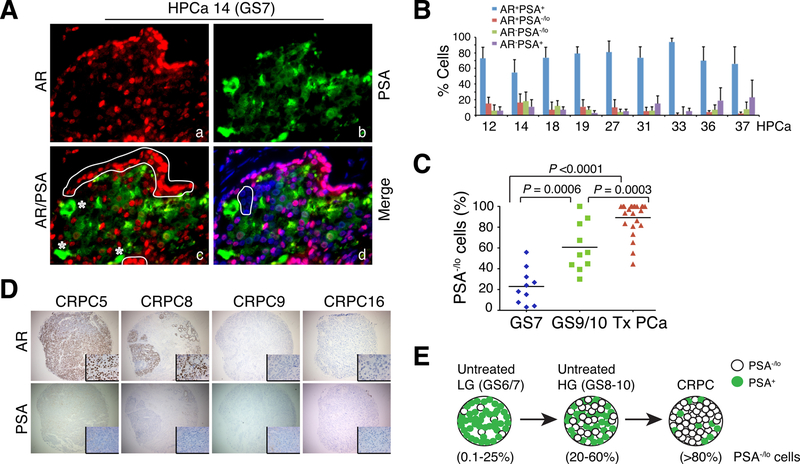
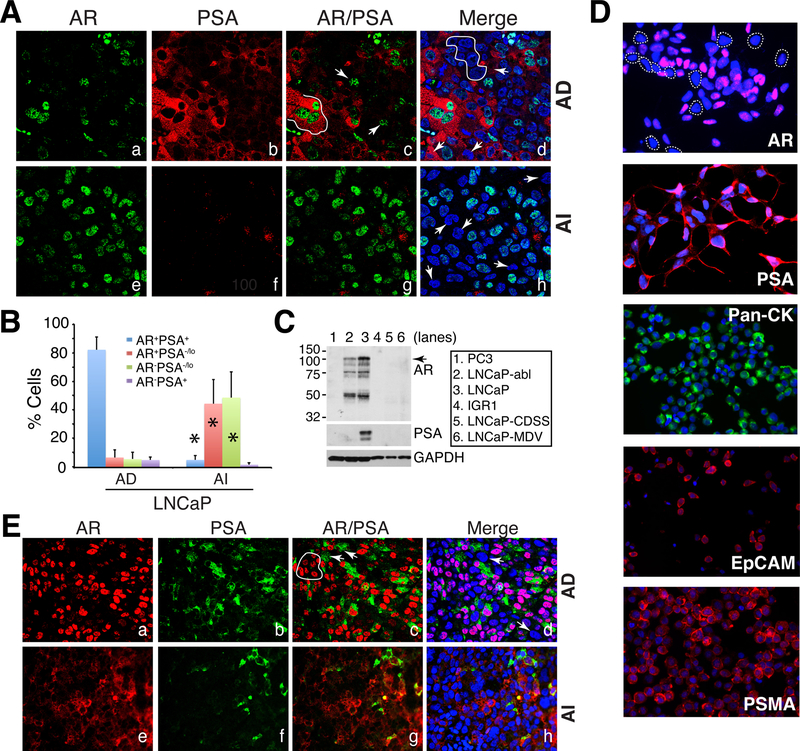
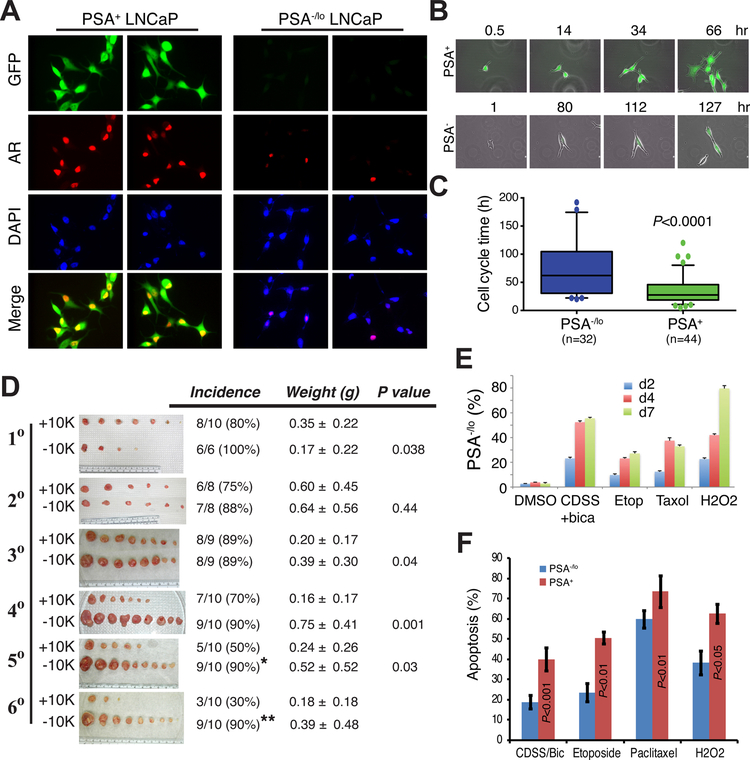
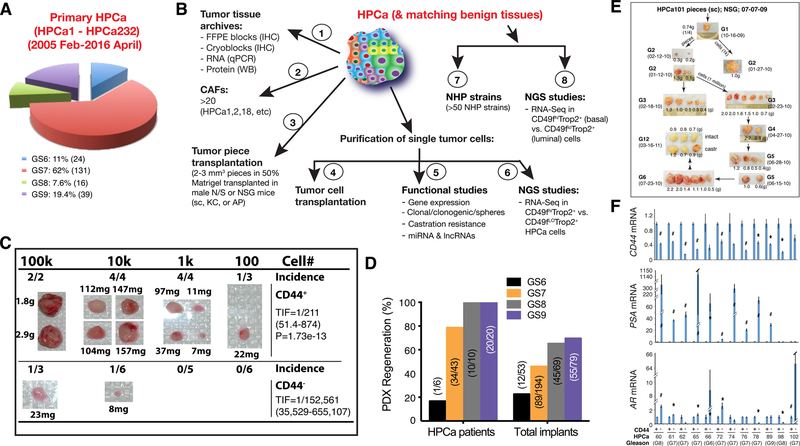
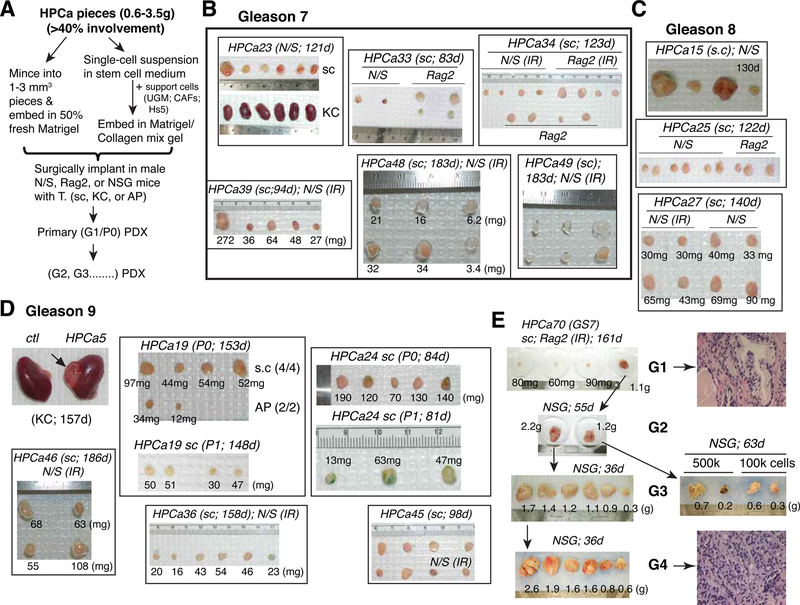
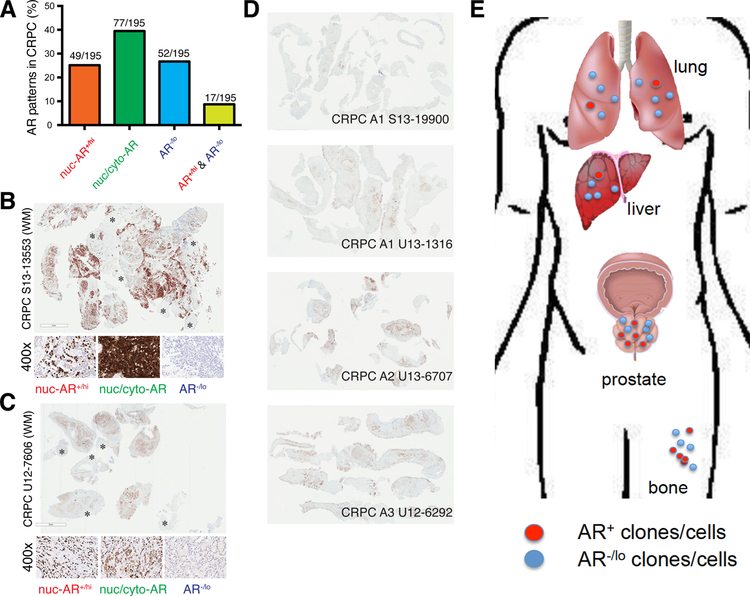
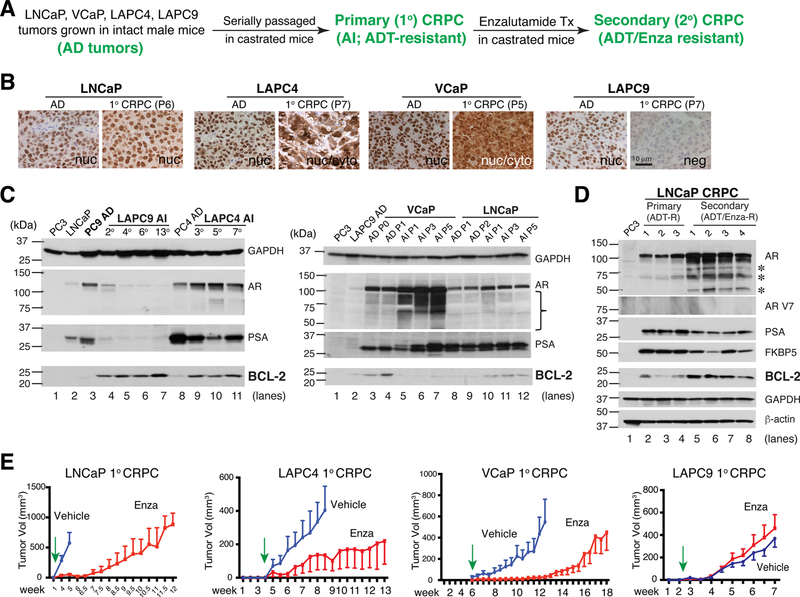
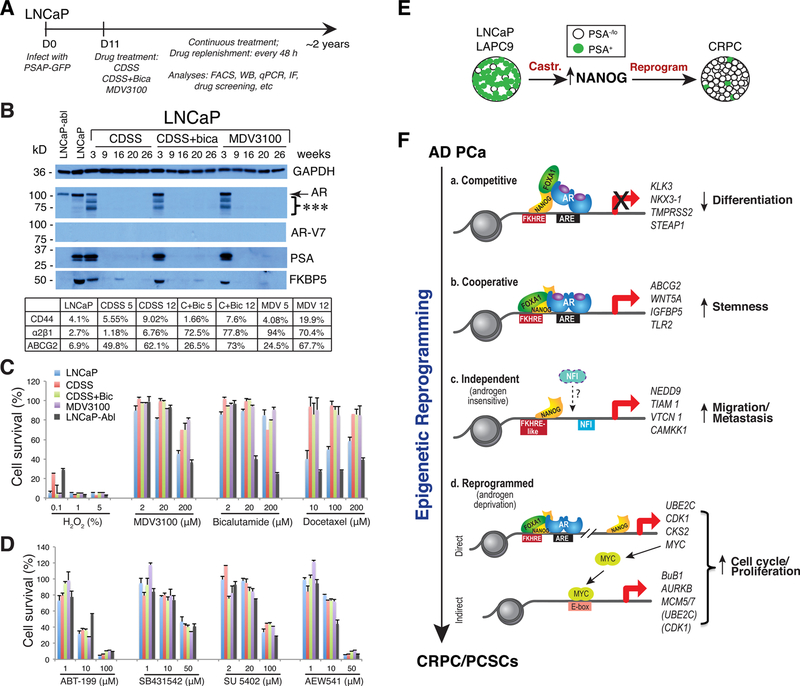
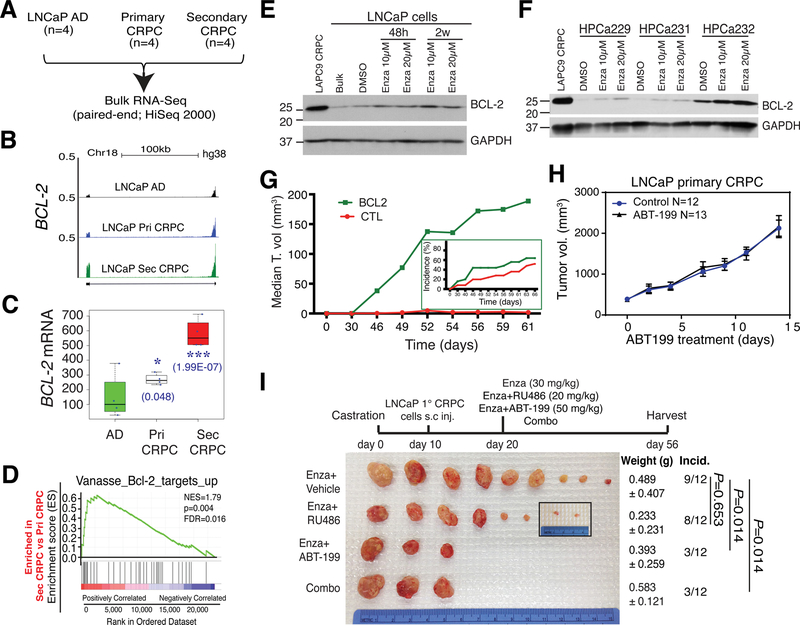
Prostate cancer (PCa) is a prevalent malignancy that occurs primarily in old males. Prostate tumors in different patients manifest significant inter-patient heterogeneity with respect to histo-morphological presentations and molecular architecture. An individual patient tumor also harbors genetically distinct clones in which PCa cells display intra-tumor heterogeneity in molecular features and phenotypic marker expression. This inherent PCa cell heterogeneity, e.g., in the expression of androgen receptor (AR), constitutes a barrier to the long-term therapeutic efficacy of AR-targeting therapies. Furthermore, tumor progression as well as therapeutic treatments induce PCa cell plasticity such that AR-positive PCa cells may turn into AR-negative cells and prostate tumors may switch lineage identity from adenocarcinomas to neuroendocrine-like tumors. This induced PCa cell plasticity similarly confers resistance to AR-targeting and other therapies. In this review, I first discuss PCa from the perspective of an abnormal organ development and deregulated cellular differentiation, and discuss the luminal progenitor cells as the likely cells of origin for PCa. I then focus on intrinsic PCa cell heterogeneity in treatment-naïve tumors with the presence of prostate cancer stem cells (PCSCs). I further elaborate on PCa cell plasticity induced by genetic alterations and therapeutic interventions, and present potential strategies to therapeutically tackle PCa cell heterogeneity and plasticity. My discussions will make it clear that, to achieve enduring clinical efficacy, both intrinsic PCa cell heterogeneity and induced PCa cell plasticity need to be targeted with novel combinatorial approaches.
Keywords: Androgen receptor; Cancer cell heterogeneity; Cancer cell plasticity; Cancer stem cells; Prostate cancer.
Copyright © 2021 The Author(s). Published by Elsevier Ltd.. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest
The authors report no declarations of interest.
Figures
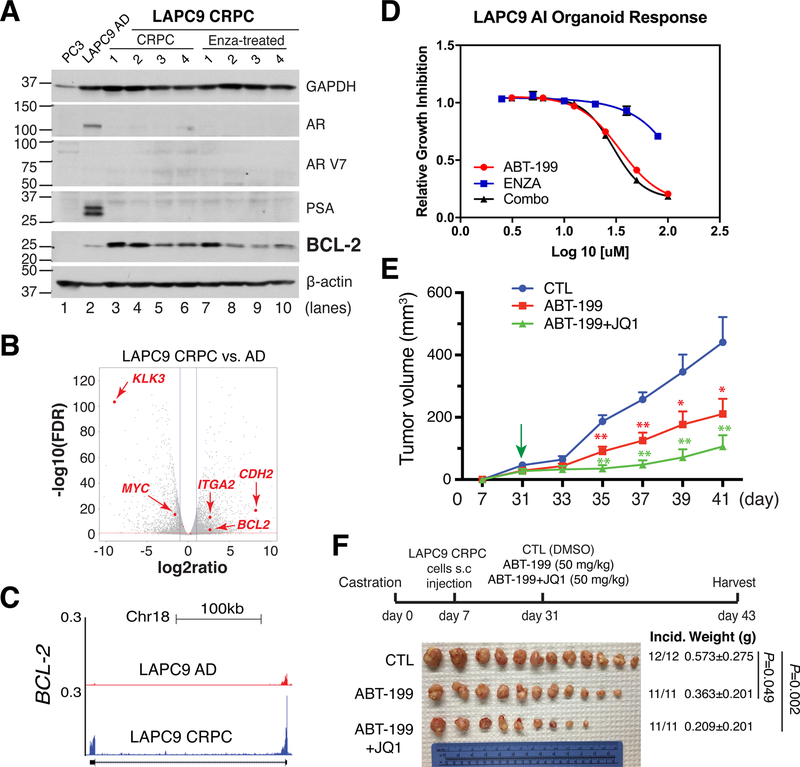
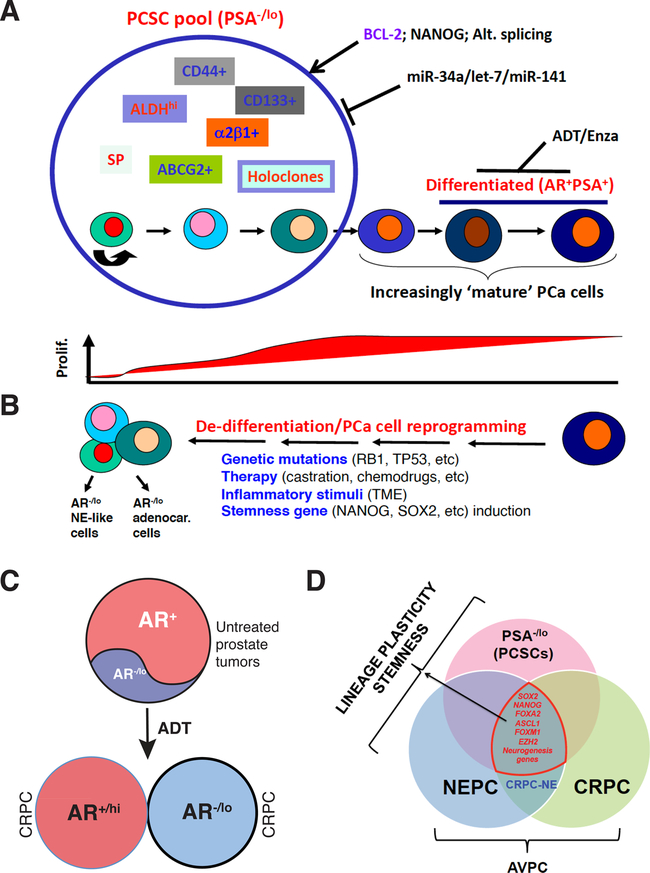
References
-
- Fenelon JC, Banerjee A, Murphy BD, Embryonic diapause: development on hold, Int. J. Dev. Biol. 58 (2014) 163–174. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
