

Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single-cell transcriptomics
- PMID: 34845188
- PMCID: PMC8630108
- DOI: 10.1038/s41467-021-26951-z
Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single-cell transcriptomics
Abstract
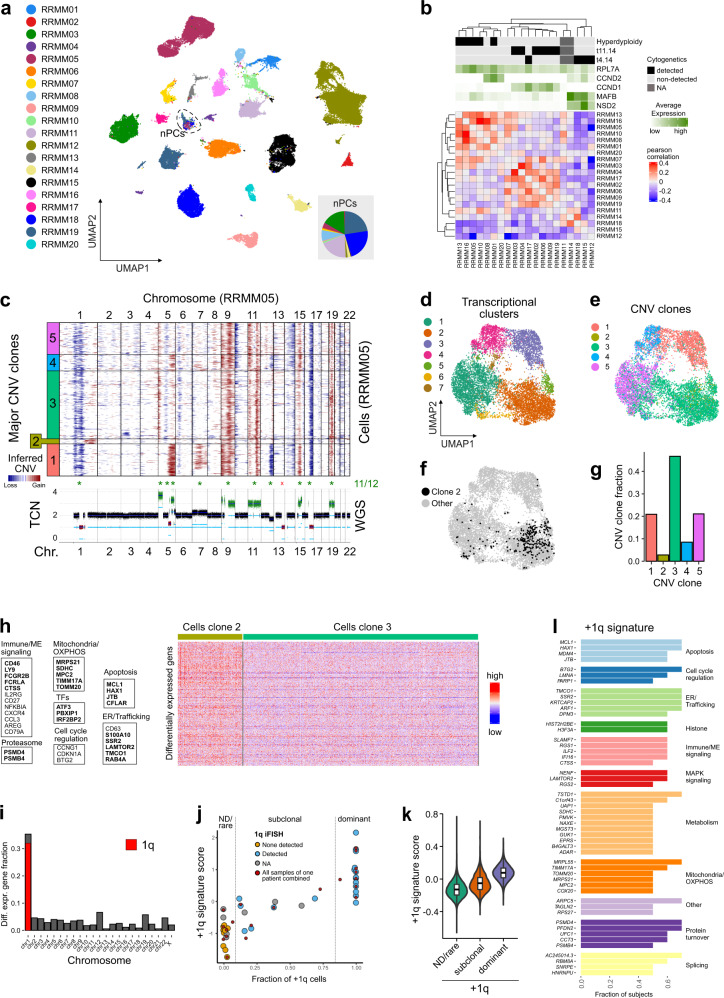
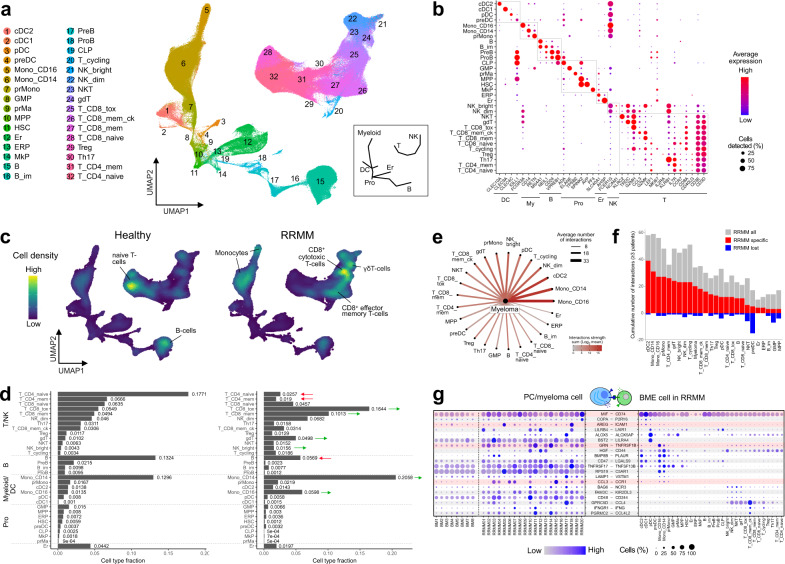
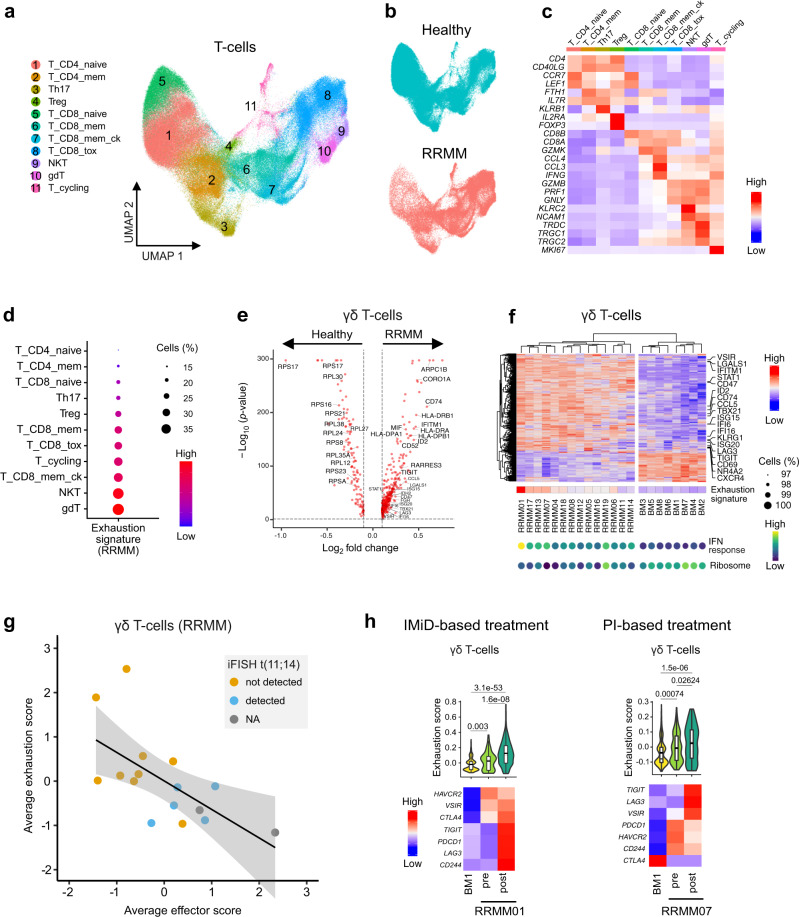
Virtually all patients with multiple myeloma become unresponsive to treatment over time. Relapsed/refractory multiple myeloma (RRMM) is accompanied by the clonal evolution of myeloma cells with heterogeneous genomic aberrations and profound changes of the bone marrow microenvironment (BME). However, the molecular mechanisms that drive drug resistance remain elusive. Here, we analyze the heterogeneous tumor cell population and its complex interaction network with the BME of 20 RRMM patients by single cell RNA-sequencing before/after treatment. Subclones with chromosome 1q-gain express a specific transcriptomic signature and frequently expand during treatment. Furthermore, RRMM cells shape an immune suppressive BME by upregulation of inflammatory cytokines and close interaction with the myeloid compartment. It is characterized by the accumulation of PD1+ γδ T-cells and tumor-associated macrophages as well as the depletion of hematopoietic progenitors. Thus, our study resolves transcriptional features of subclones in RRMM and mechanisms of microenvironmental reprogramming with implications for clinical decision-making.
© 2021. The Author(s).
Conflict of interest statement
H.G. – Grants/provisions: Amgen, BMS, Celgene, Chugai, Janssen, Johns Hopkins University, Sanofi; Research support: Amgen, BMS, Celgene, Chugai, Janssen, Incyte, Molecular Partners, Merck Sharp and Dohme (MSD), Sanofi, Mundipharma GmbH, Takeda, Novartis; Advisory Boards: Adaptive Biotechnology, Amgen, BMS, Celgene, Janssen, Sanofi, Takeda; Honoraria: Amgen, BMS, Celgene, Chugai, GlaxoSmithKline (GSK), Janssen, Novartis, Sanofi. C.M.T. – Grants/provisions: Pfizer, Daiichi Sankyo, BiolineRx; Research support: Abbvie, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eisai, Fresenius, Gilead, Hexal, Janssen, Jazz Pharmaceuticals, MSD, Novartis, Pharmamar, Pfizer, Roche, Shire, Takeda, Affimed; Advisory Boards: Pfizer, Janssen. The other authors declare no competing interests.
Figures




References
-
- Kumar SK, et al. Multiple myeloma. Nat. Rev. Dis. Prim. 2017;3:17046. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
