

Mutant IDH Inhibits IFNγ-TET2 Signaling to Promote Immunoevasion and Tumor Maintenance in Cholangiocarcinoma
- PMID: 34848557
- PMCID: PMC8904298
- DOI: 10.1158/2159-8290.CD-21-1077
Mutant IDH Inhibits IFNγ-TET2 Signaling to Promote Immunoevasion and Tumor Maintenance in Cholangiocarcinoma
Abstract
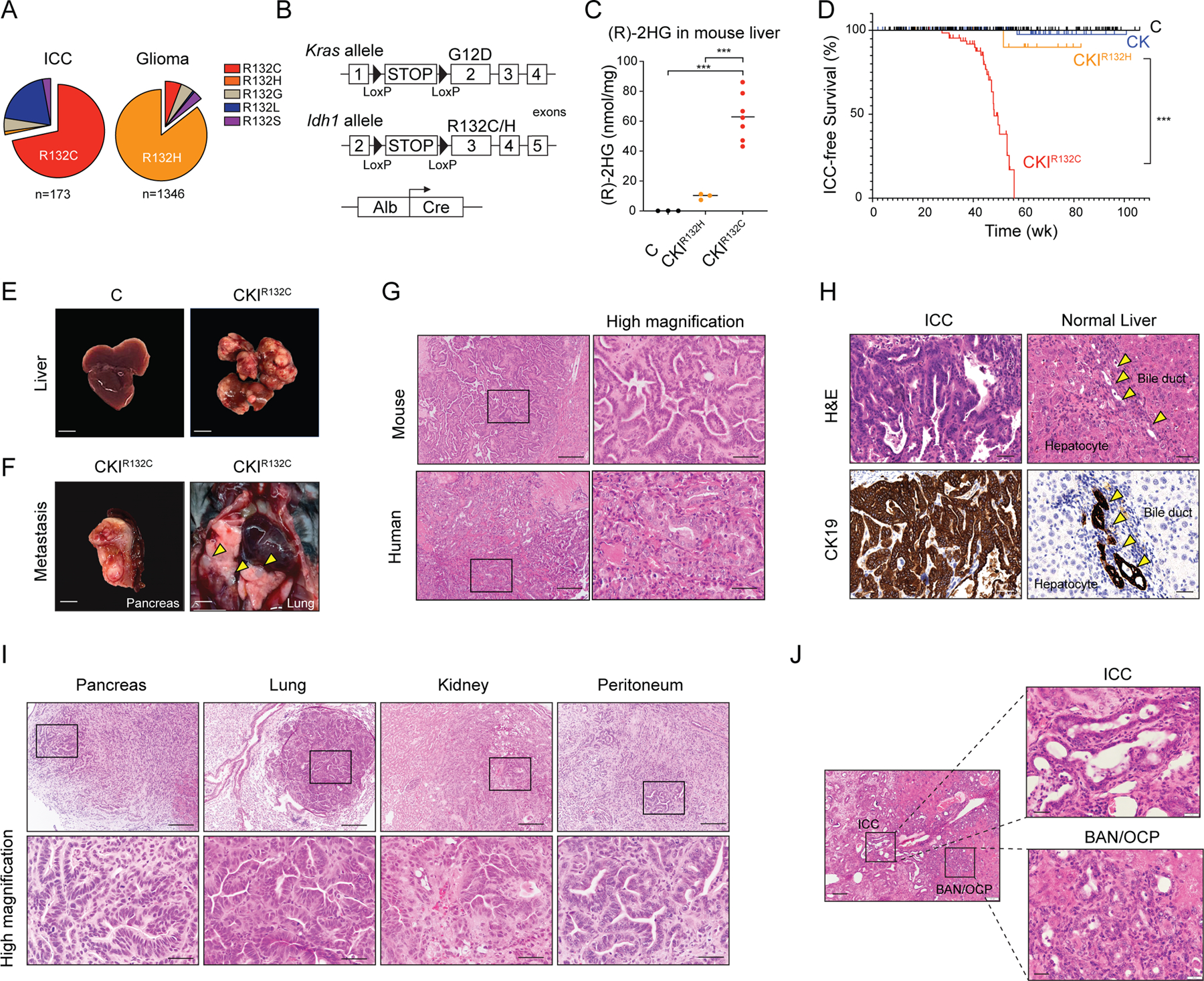
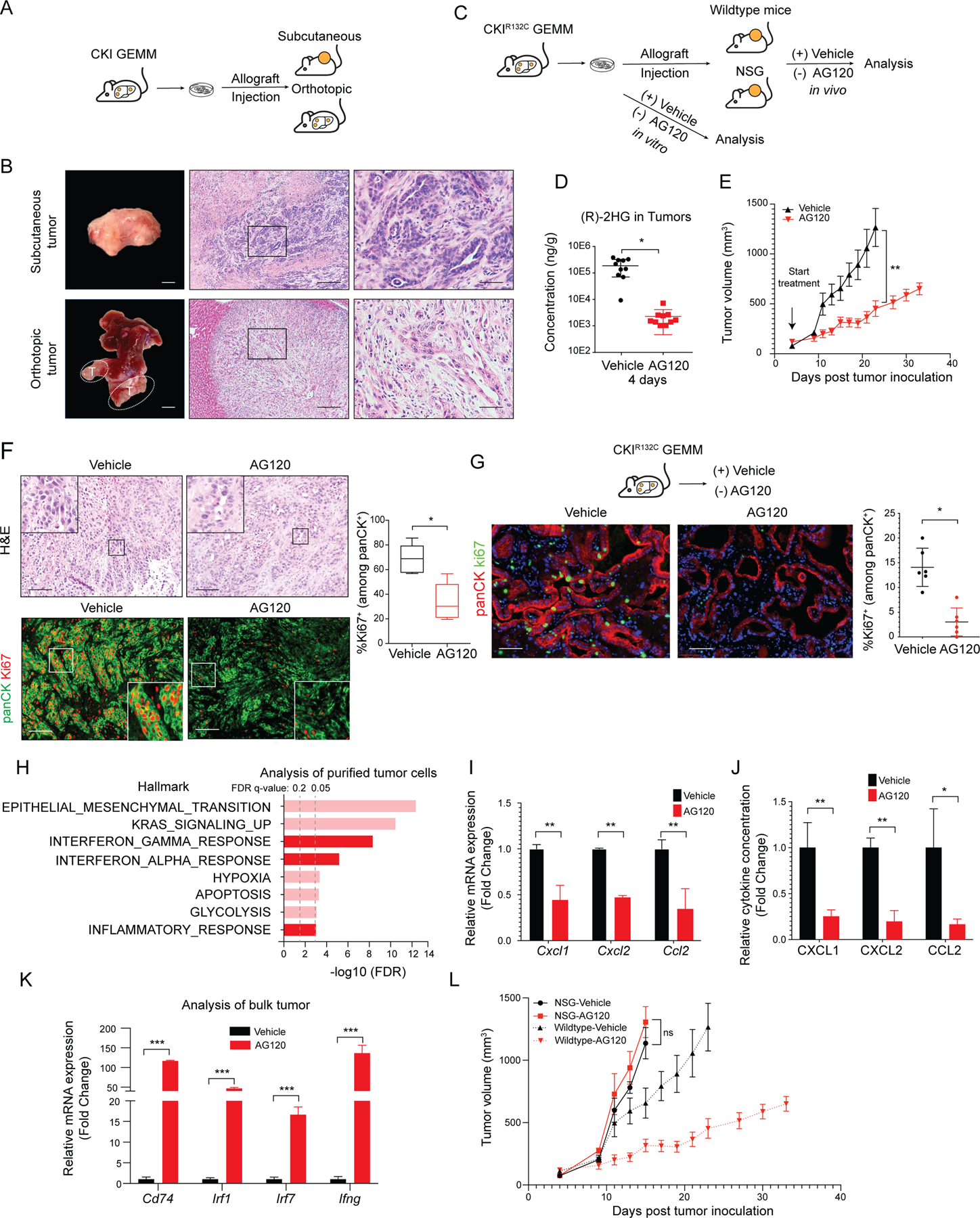
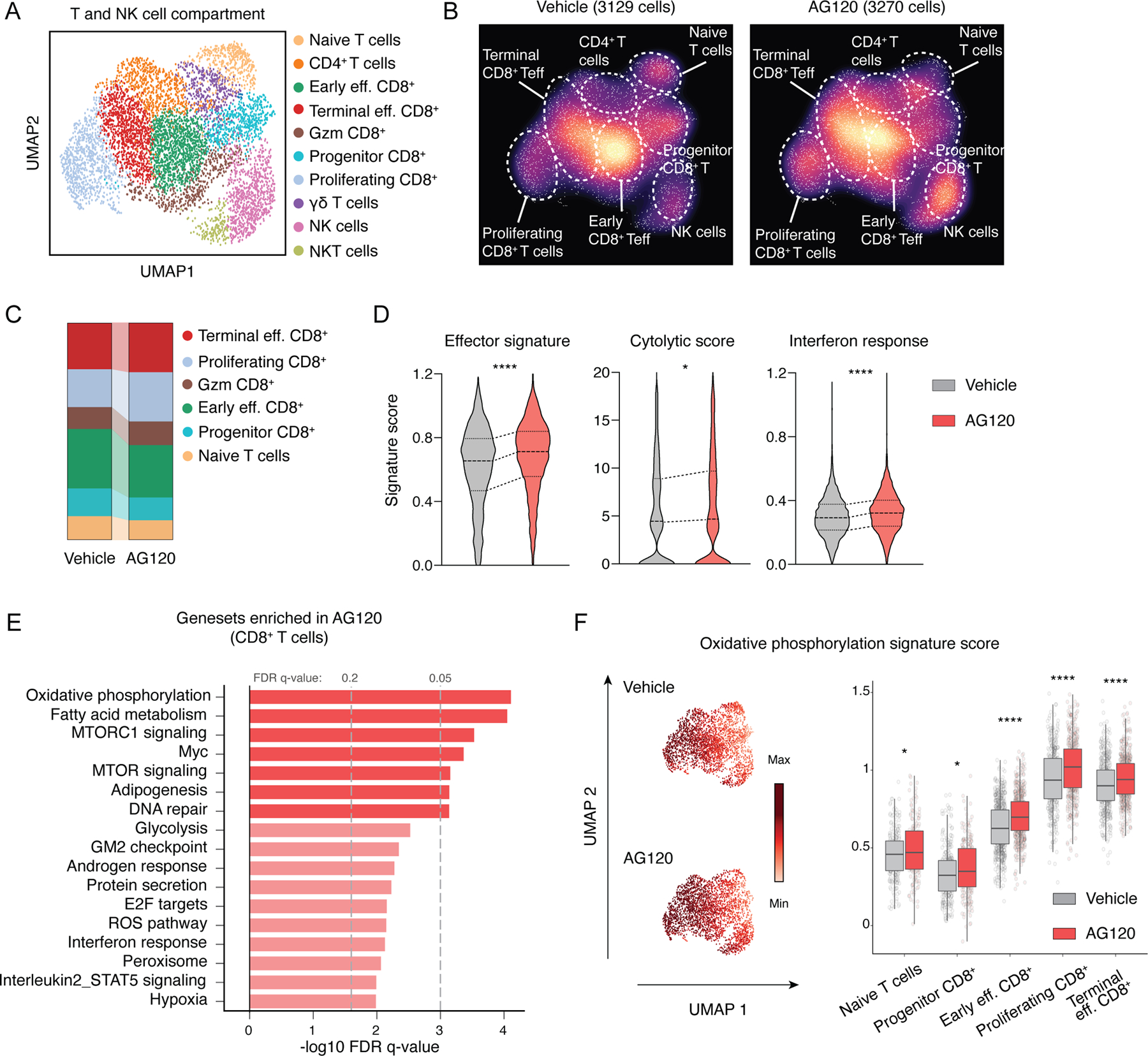
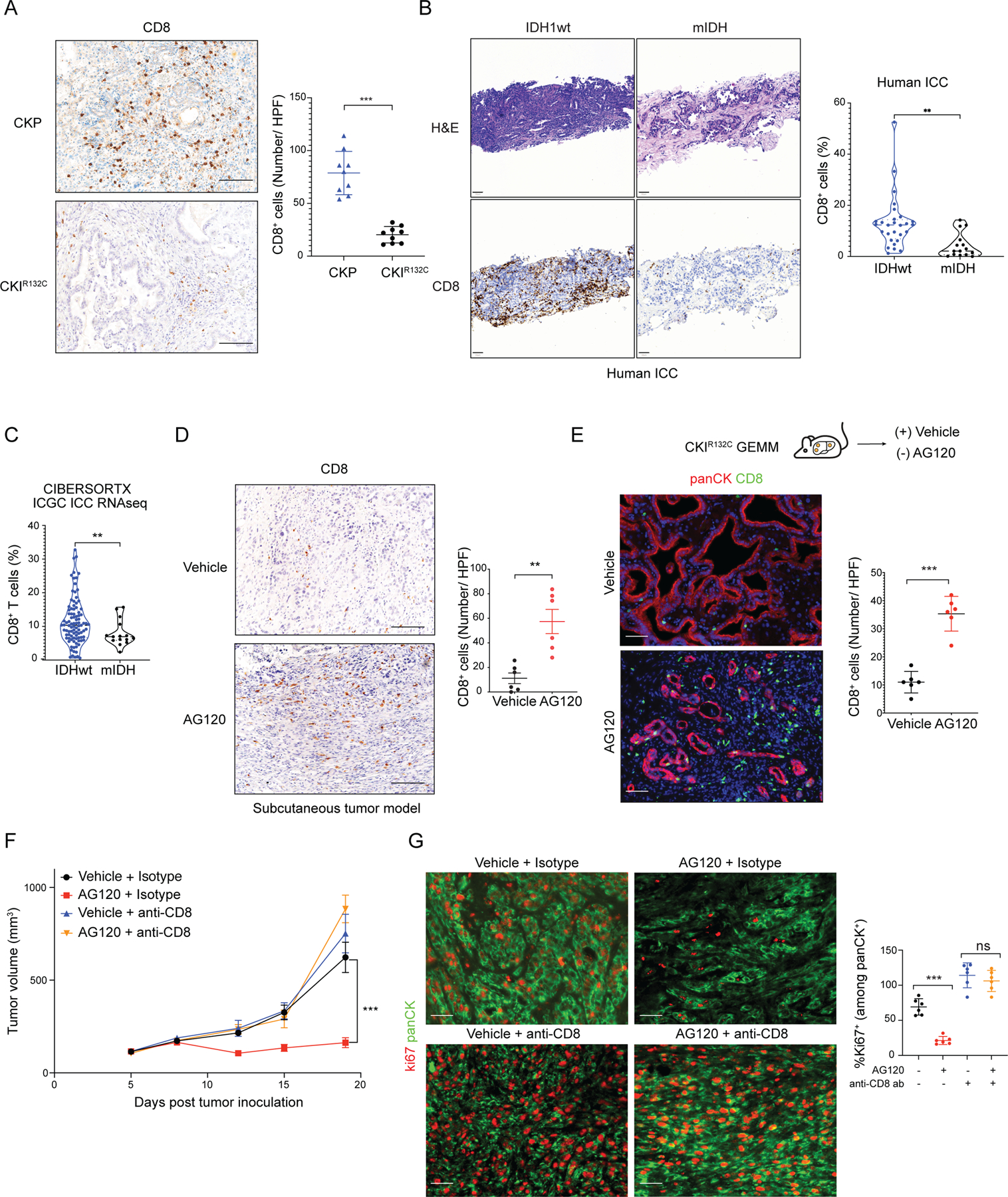
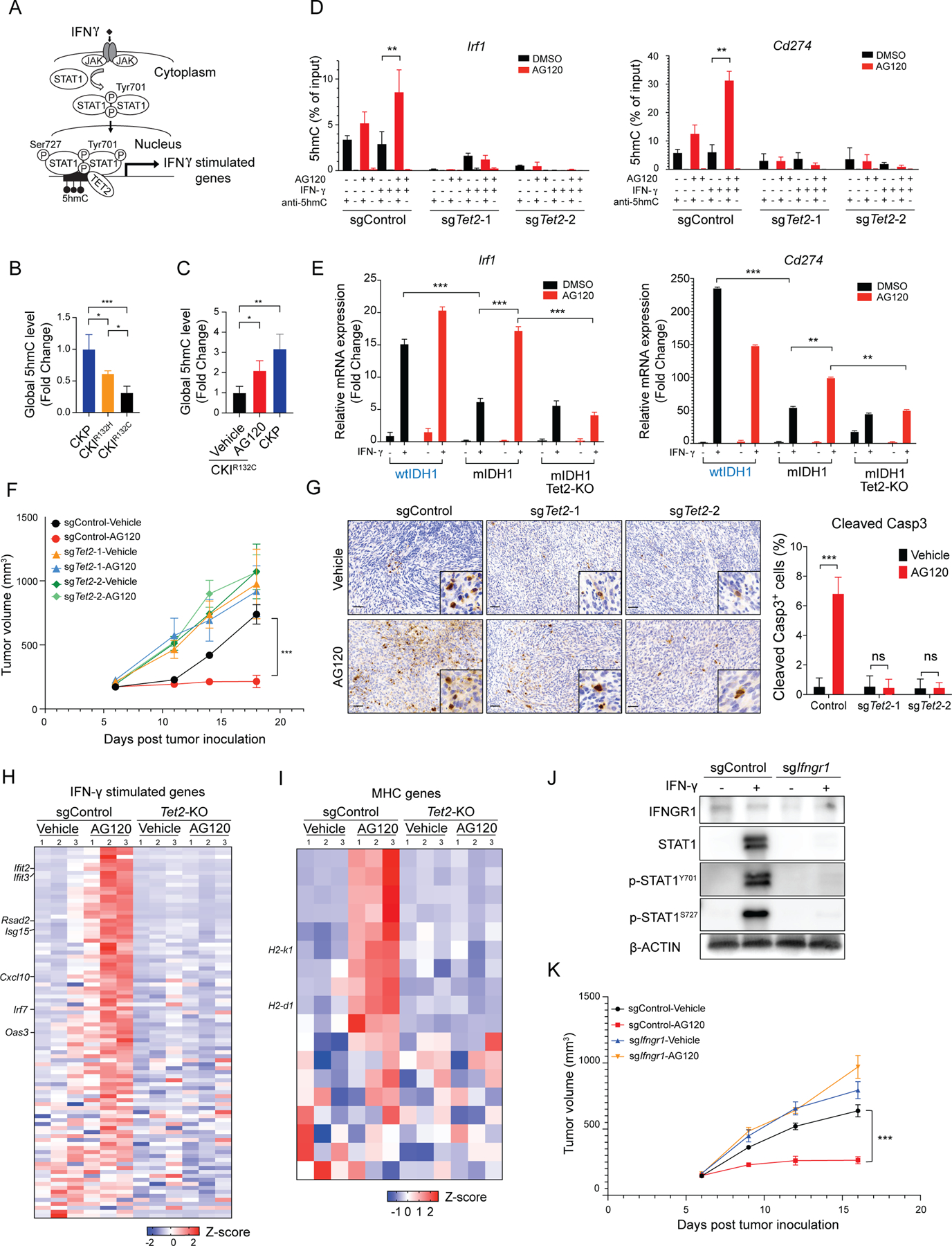
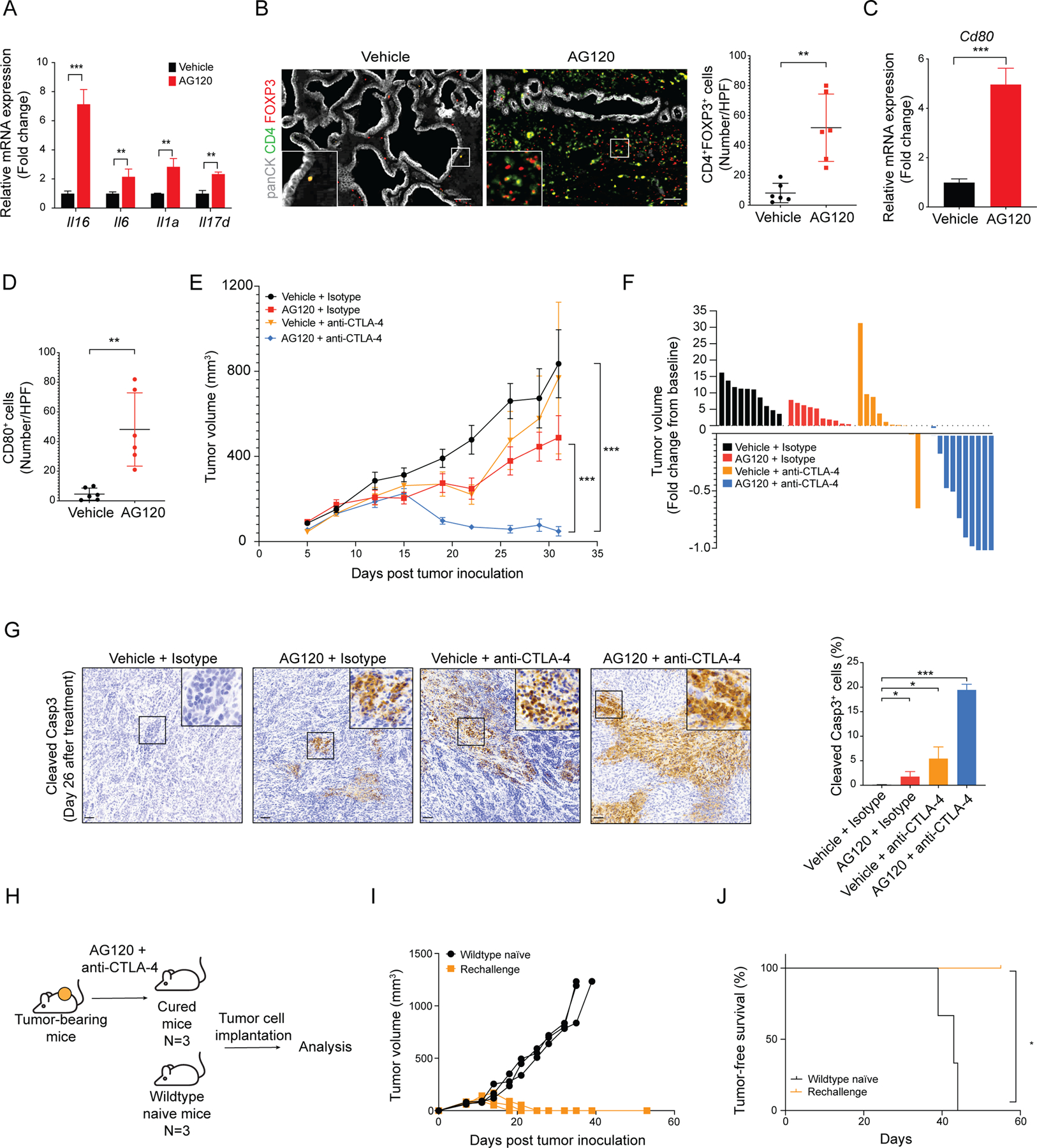
Isocitrate dehydrogenase 1 mutations (mIDH1) are common in cholangiocarcinoma. (R)-2-hydroxyglutarate generated by the mIDH1 enzyme inhibits multiple α-ketoglutarate-dependent enzymes, altering epigenetics and metabolism. Here, by developing mIDH1-driven genetically engineered mouse models, we show that mIDH1 supports cholangiocarcinoma tumor maintenance through an immunoevasion program centered on dual (R)-2-hydroxyglutarate-mediated mechanisms: suppression of CD8+ T-cell activity and tumor cell-autonomous inactivation of TET2 DNA demethylase. Pharmacologic mIDH1 inhibition stimulates CD8+ T-cell recruitment and interferon γ (IFNγ) expression and promotes TET2-dependent induction of IFNγ response genes in tumor cells. CD8+ T-cell depletion or tumor cell-specific ablation of TET2 or IFNγ receptor 1 causes treatment resistance. Whereas immune-checkpoint activation limits mIDH1 inhibitor efficacy, CTLA4 blockade overcomes immunosuppression, providing therapeutic synergy. The findings in this mouse model of cholangiocarcinoma demonstrate that immune function and the IFNγ-TET2 axis are essential for response to mIDH1 inhibition and suggest a novel strategy for potentiating efficacy.
Significance: Mutant IDH1 inhibition stimulates cytotoxic T-cell function and derepression of the DNA demethylating enzyme TET2, which is required for tumor cells to respond to IFNγ. The discovery of mechanisms of treatment efficacy and the identification of synergy by combined CTLA4 blockade provide the foundation for new therapeutic strategies. See related commentary by Zhu and Kwong, p. 604. This article is highlighted in the In This Issue feature, p. 587.
©2021 American Association for Cancer Research.
Figures
Comment in
-
IDH1 Inhibition Reawakens the Immune Response against Cholangiocarcinoma.Cancer Discov. 2022 Mar 1;12(3):604-605. doi: 10.1158/2159-8290.CD-21-1643. Cancer Discov. 2022. PMID: 35257150
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
