

A 1-year and 4-month-old child with mucopolysaccharidoses type II: A clinical case report from Ethiopia
- PMID: 34849229
- PMCID: PMC8607870
- DOI: 10.1002/ccr3.5122
A 1-year and 4-month-old child with mucopolysaccharidoses type II: A clinical case report from Ethiopia
Abstract
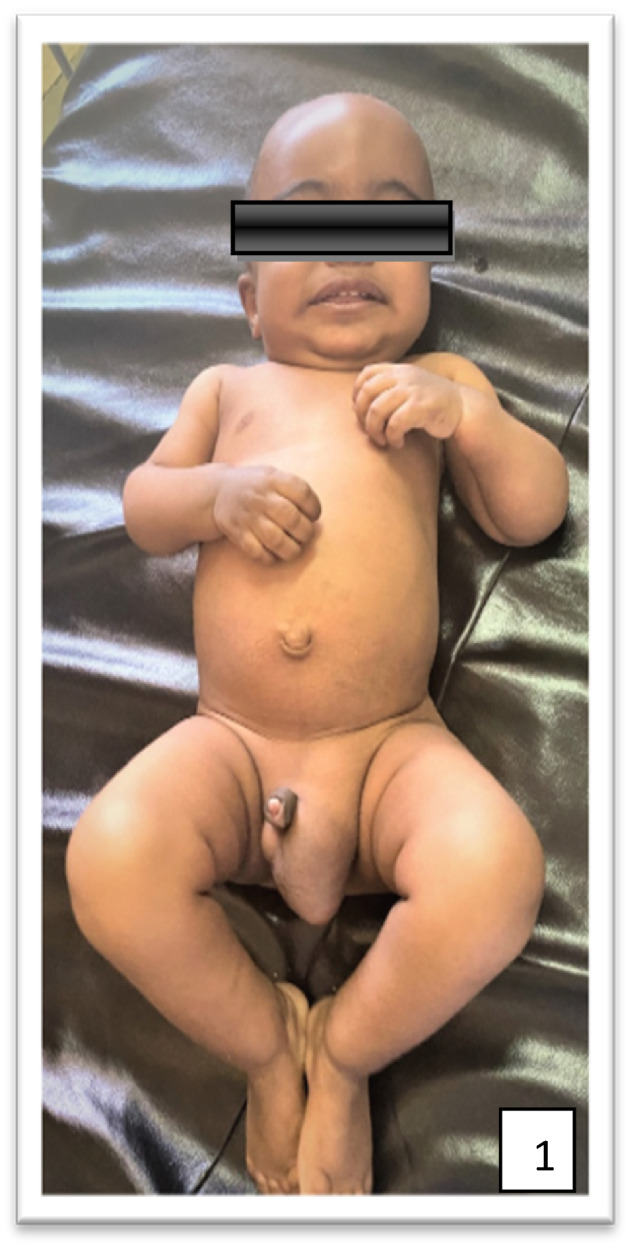
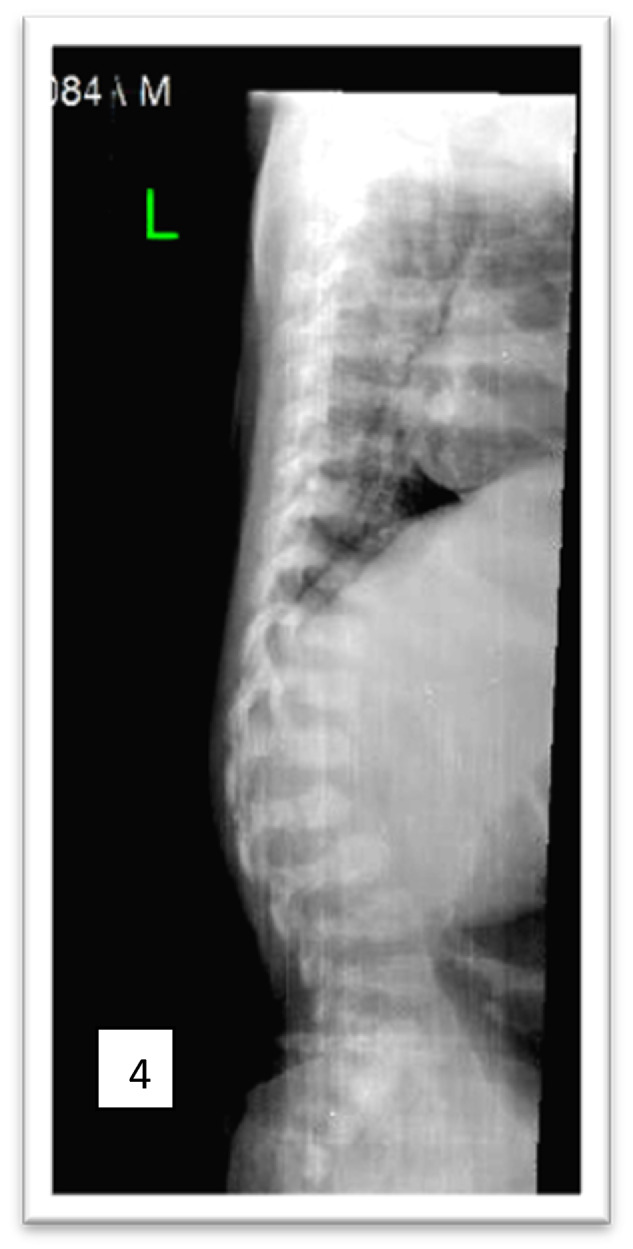
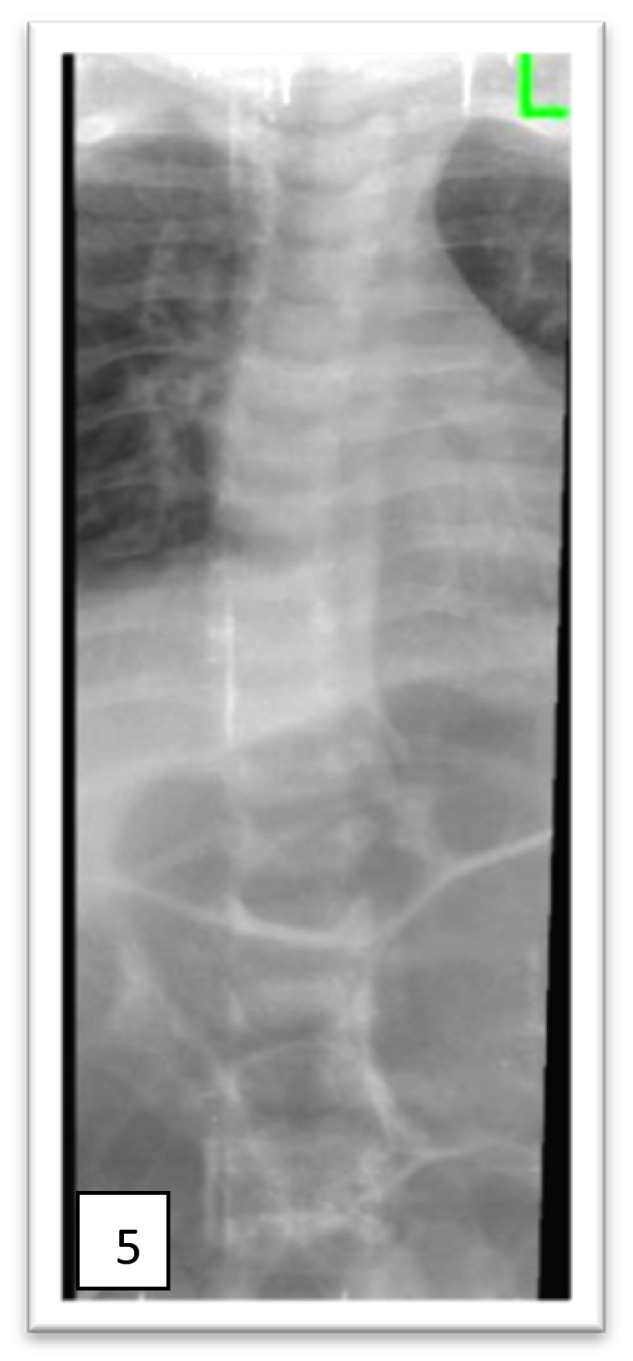
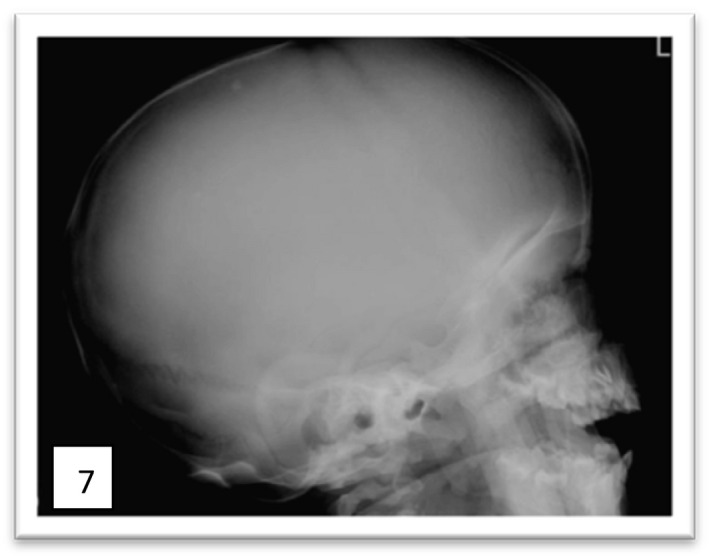
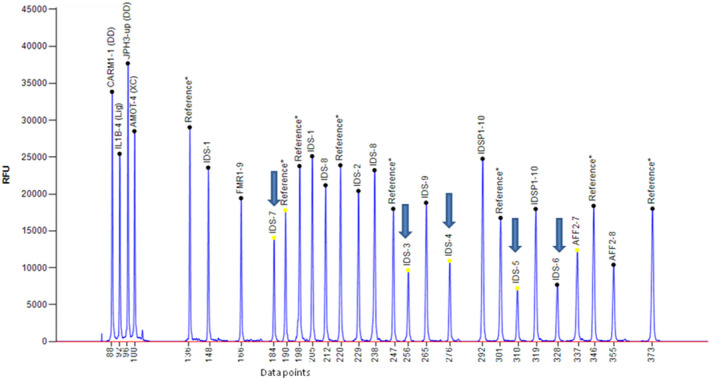
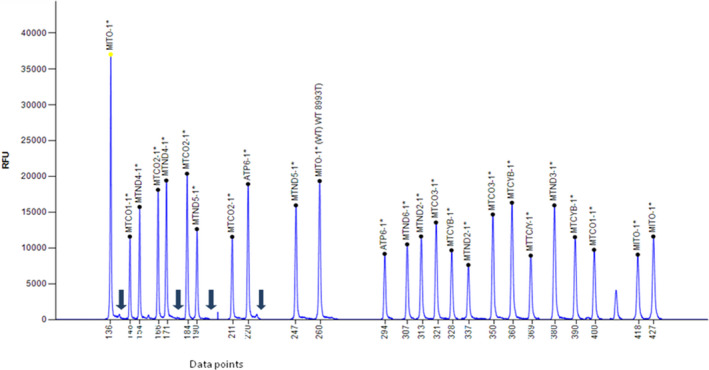
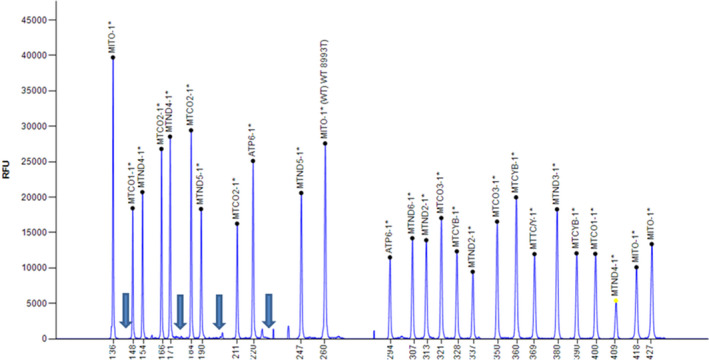
Mucopolysaccharidoses (MPSs) are a class of lysosomal storage disorders resulting in progressive disease manifestations and are caused by pathogenic variants in genes coding for enzymes needed to degrade glycosaminoglycans. While most of the seven MPSs are autosomal recessive disorders, MPS II, also known as Hunter syndrome, is inherited in an X-linked recessive manner and is the most common MPS. Here, we report a 1-year and 4-month-old boy who presented with delayed developmental milestones, back deformity, and left scrotal swelling noticed by parents at one year of age. He has coarse facial appearance with macrocephaly, widened wrists, congenital dermal melanocytosis on his back, kyphotic deformity in the thoracolumbar area and left-sided inguinal hernia all consistent with a suspected MPS II diagnosis. The MPS II diagnosis was subsequently confirmed with genetic testing of the IDS gene. To our knowledge, this is the first case of MPS II reported from Ethiopia. This case shows the importance of early clinical recognition of genetic conditions and the utility of genetic testing for confirmation. The diagnosis provided important surveillance and natural history information for the patient's providers and family.
Keywords: Hunter syndrome; glycosaminoglycans; iduronate‐2‐sulfatase; mucopolysaccharidosis.
© 2021 The Authors. Clinical Case Reports published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors do not have any conflict of interest.
Figures




References
-
- Jürgen W S. Mucopolysaccharidoses. In: Robert M K, Joseph W st G, Nathan J B, Samir S S, Robert C T, Karen M W, Richard E B, eds. Nelson Text Book of Pediatrics. Elsevier; 2016: 1551‐1552.
-
- Germaine LD. Hunter syndrome (mucopolysaccharidosis type II): background, pathophysiology, epidemiology. EMedicine, 4 2018, emedicine.medscape.com/article/944723‐overview#:~:text=In%201917%20at%20.... Accessed February 5, 2021.
-
- Burton BK, Jego V, Mikl J, Jones SA. Survival in idursulfase‐treated and untreated patients with mucopolysaccharidosis type II: data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis. 2017;40(6):867‐874. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
