

Calcific aortic valve disease: from molecular and cellular mechanisms to medical therapy
- PMID: 34849696
- PMCID: PMC8843796
- DOI: 10.1093/eurheartj/ehab757
Calcific aortic valve disease: from molecular and cellular mechanisms to medical therapy
Abstract
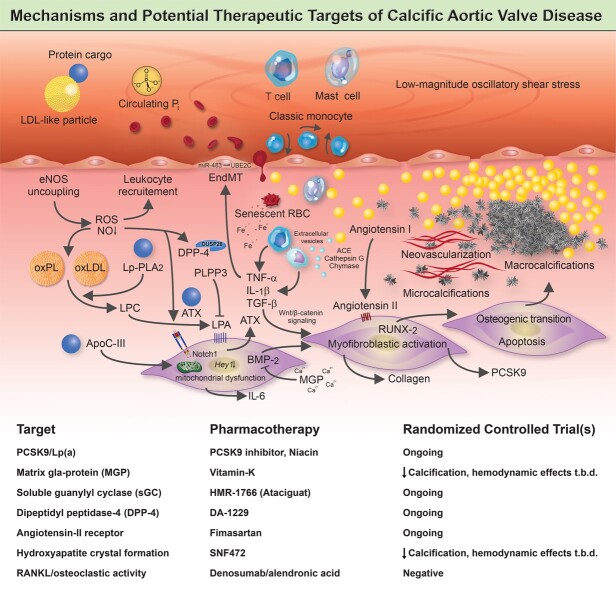
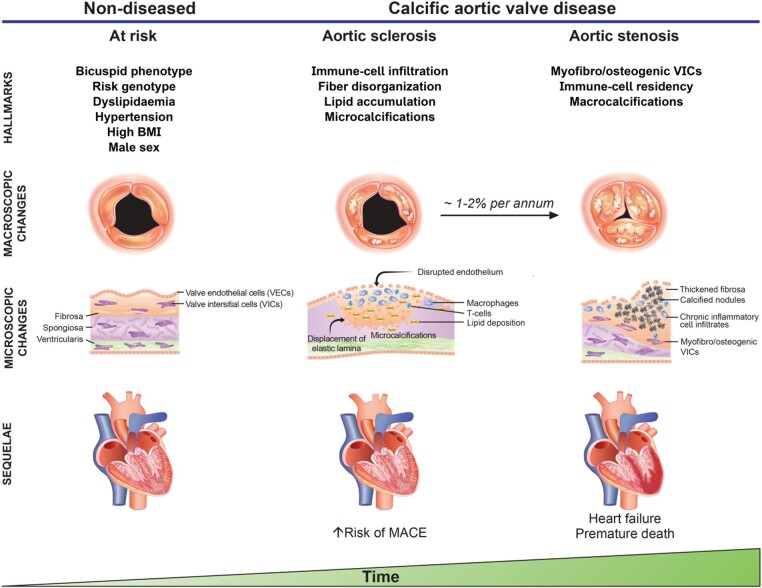
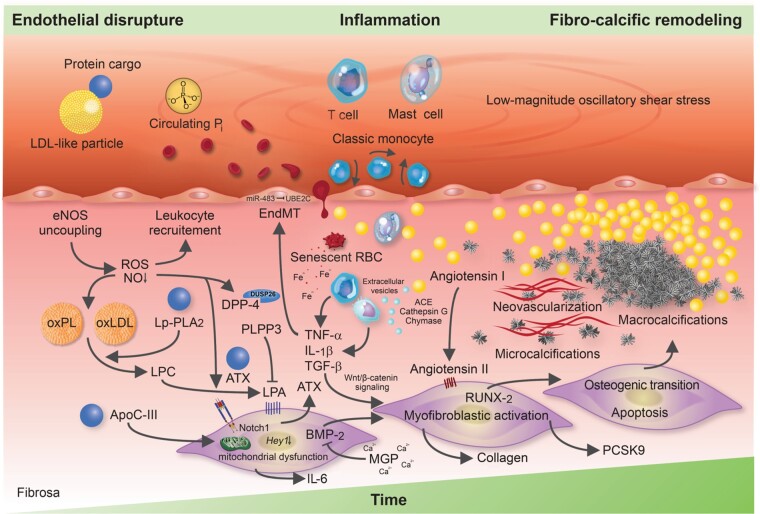
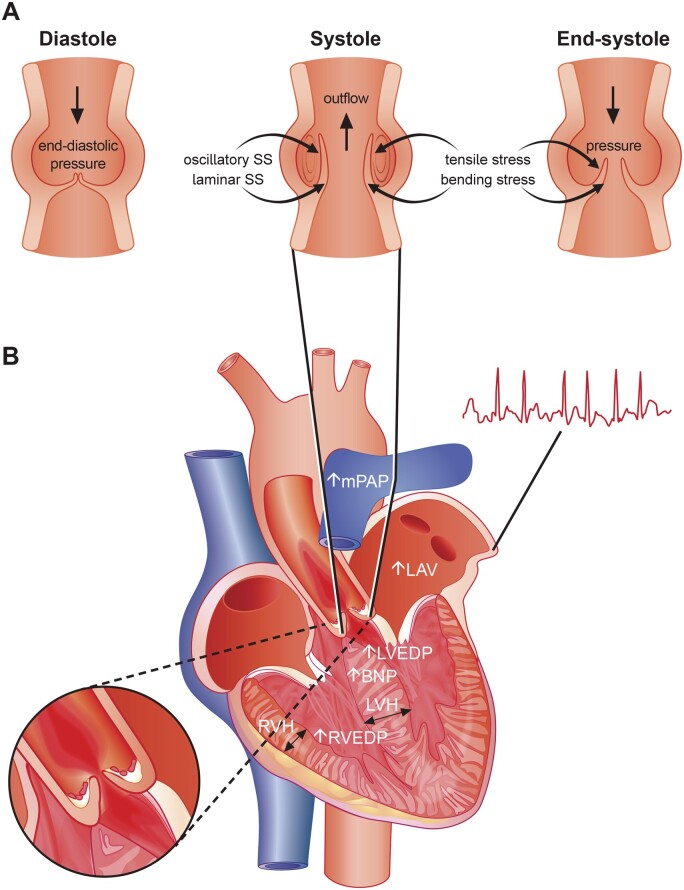
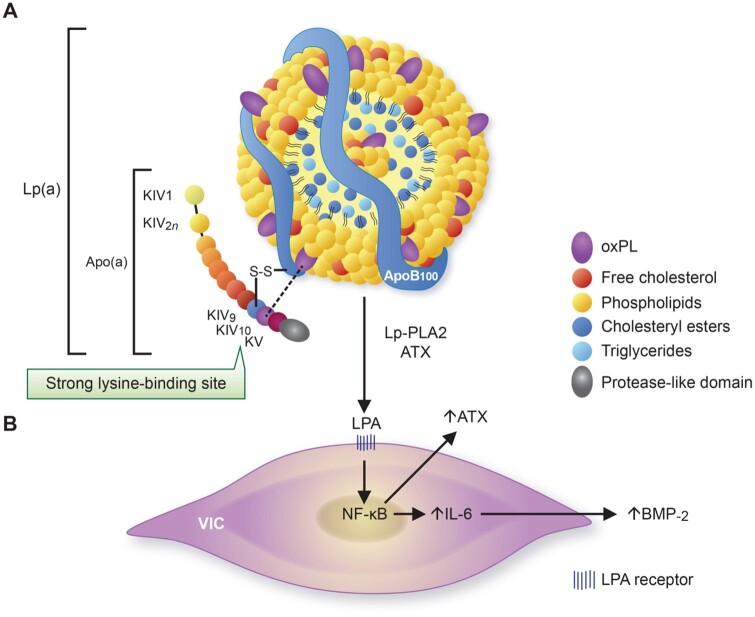
Calcific aortic valve disease (CAVD) is a highly prevalent condition that comprises a disease continuum, ranging from microscopic changes to profound fibro-calcific leaflet remodelling, culminating in aortic stenosis, heart failure, and ultimately premature death. Traditional risk factors, such as hypercholesterolaemia and (systolic) hypertension, are shared among atherosclerotic cardiovascular disease and CAVD, yet the molecular and cellular mechanisms differ markedly. Statin-induced low-density lipoprotein cholesterol lowering, a remedy highly effective for secondary prevention of atherosclerotic cardiovascular disease, consistently failed to impact CAVD progression or to improve patient outcomes. However, recently completed phase II trials provide hope that pharmaceutical tactics directed at other targets implicated in CAVD pathogenesis offer an avenue to alter the course of the disease non-invasively. Herein, we delineate key players of CAVD pathobiology, outline mechanisms that entail compromised endothelial barrier function, and promote lipid homing, immune-cell infiltration, and deranged phospho-calcium metabolism that collectively perpetuate a pro-inflammatory/pro-osteogenic milieu in which valvular interstitial cells increasingly adopt myofibro-/osteoblast-like properties, thereby fostering fibro-calcific leaflet remodelling and eventually resulting in left ventricular outflow obstruction. We provide a glimpse into the most promising targets on the horizon, including lipoprotein(a), mineral-binding matrix Gla protein, soluble guanylate cyclase, dipeptidyl peptidase-4 as well as candidates involved in regulating phospho-calcium metabolism and valvular angiotensin II synthesis and ultimately discuss their potential for a future therapy of this insidious disease.
Keywords: Ageing; Calcific aortic valve disease; Lipoprotein(a); Medical therapy; Nitric oxide; Notch1.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author(s) 2021. For permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Novaro GM, Katz R, Aviles RJ et al. Clinical factors, but not C-reactive protein, predict progression of calcific aortic-valve disease. The Cardiovascular Health Study. J Am Coll Cardiol 2007;50:1992–1998. - PubMed
-
- Sverdlov AL, Ngo DTM, Chan WPA et al. Determinants of aortic sclerosis progression: implications regarding impairment of nitric oxide signalling and potential therapeutics. Eur Heart J 2012;33:2419–2425. - PubMed
-
- Faggiano P, Antonini-Canterin F, Erlicher A et al. Progression of aortic valve sclerosis to aortic stenosis. Am J Cardiol 2003;91:99–101. - PubMed
-
- Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med 1999;341:142–147. - PubMed
-
- Coffey S, Cox B, Williams MJA. The prevalence, incidence, progression, and risks of aortic valve sclerosis: a systematic review and meta-analysis. J Am Coll Cardiol 2014;63:2852–2861. - PubMed
