

Assessing genome-wide dynamic changes in enhancer activity during early mESC differentiation by FAIRE-STARR-seq
- PMID: 34850108
- PMCID: PMC8643627
- DOI: 10.1093/nar/gkab1100
Assessing genome-wide dynamic changes in enhancer activity during early mESC differentiation by FAIRE-STARR-seq
Abstract
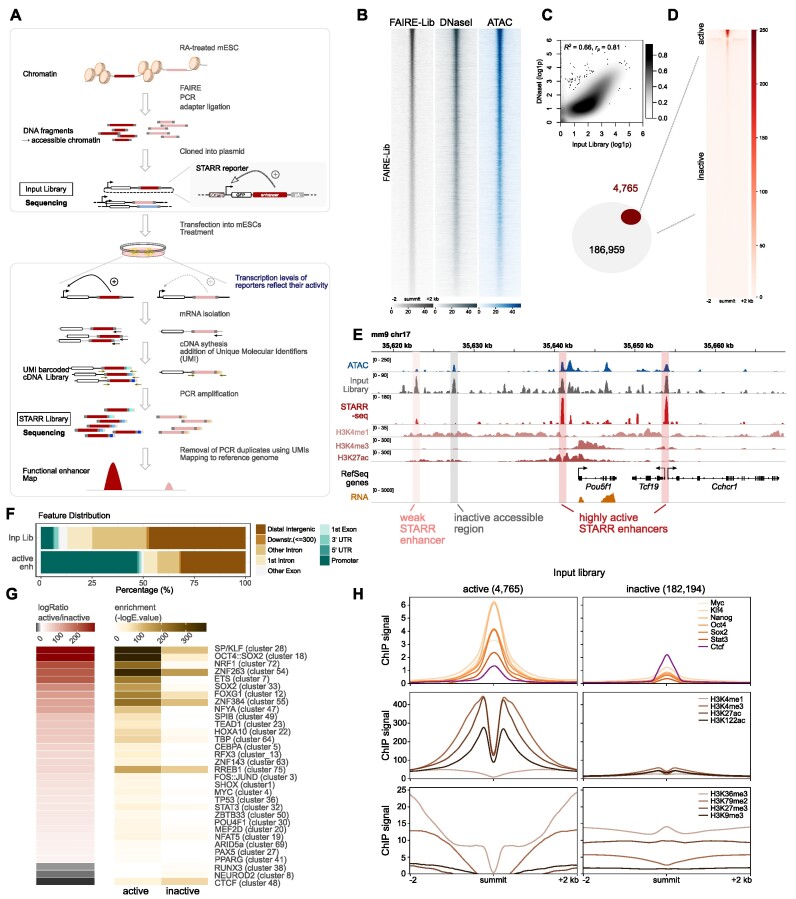
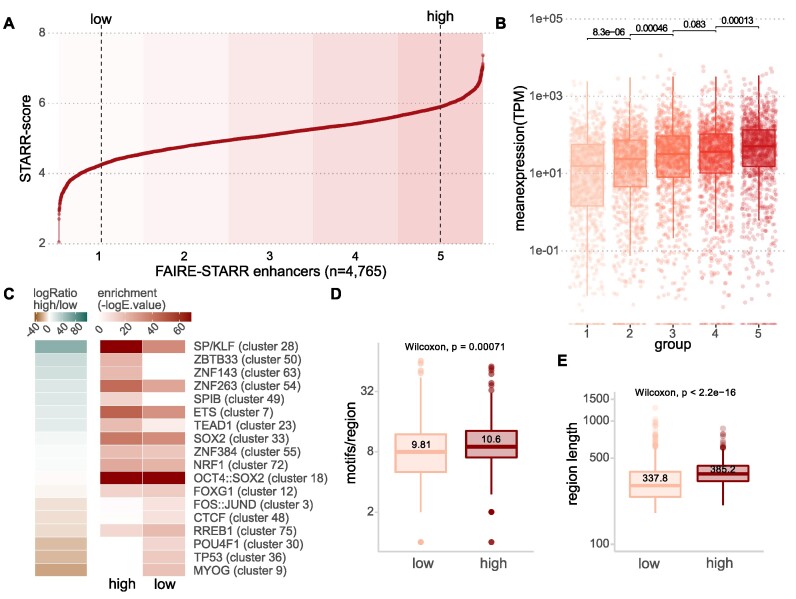
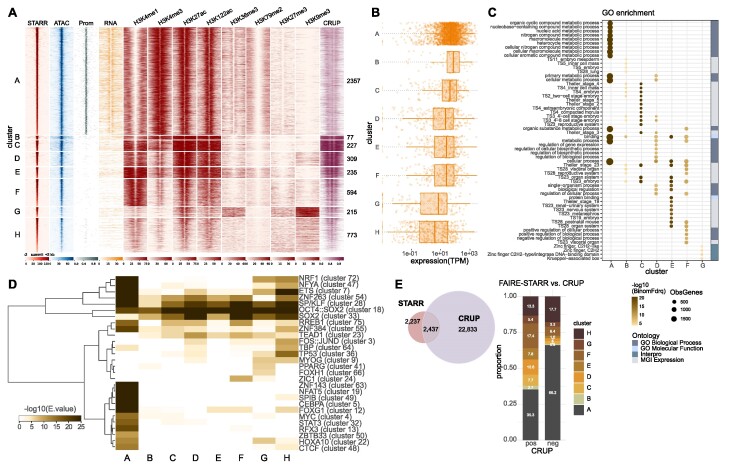
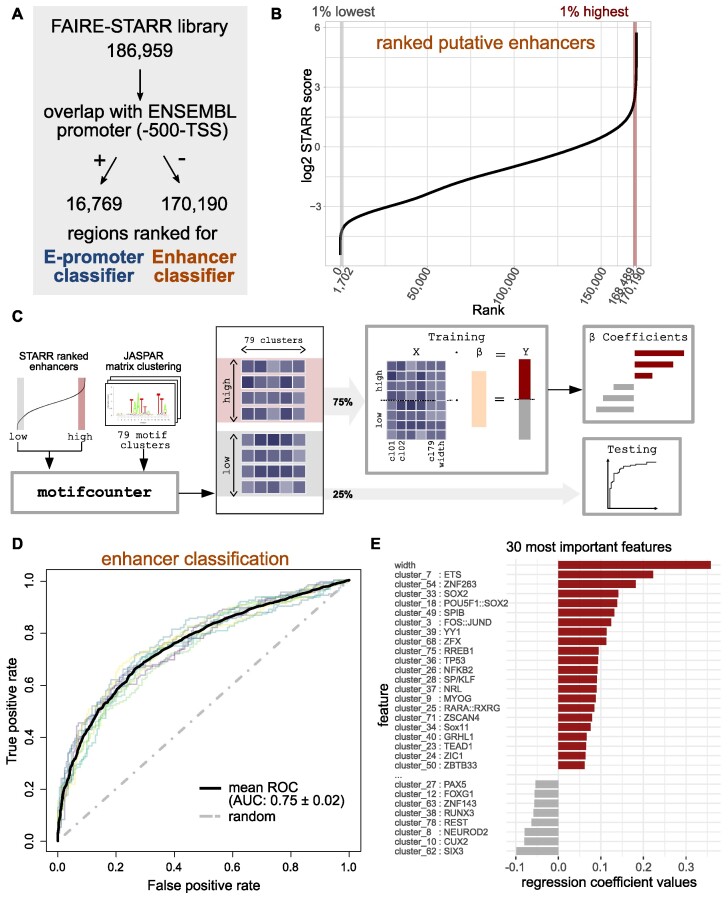
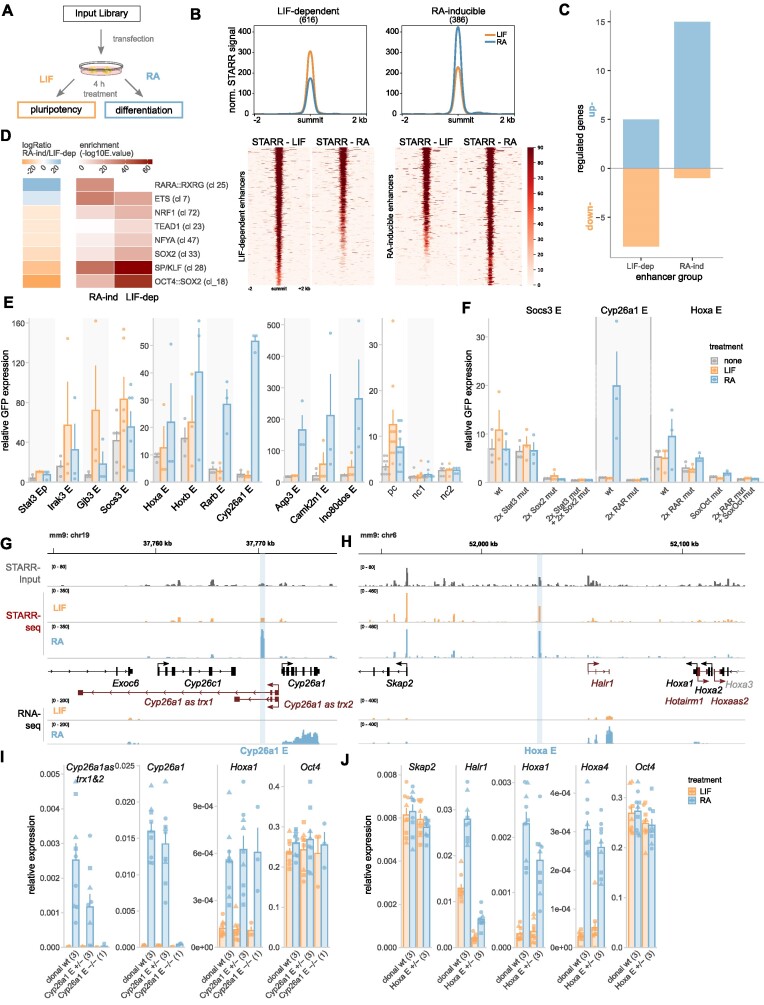
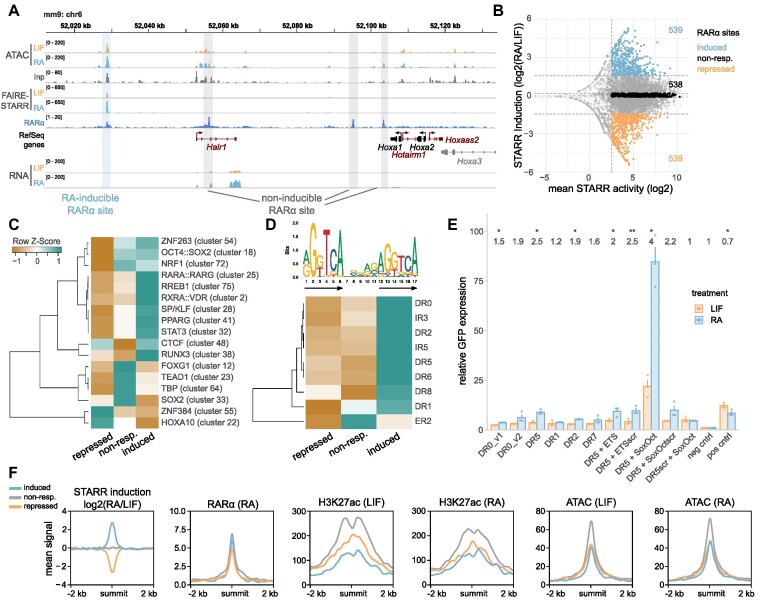
Embryonic stem cells (ESCs) can differentiate into any given cell type and therefore represent a versatile model to study the link between gene regulation and differentiation. To quantitatively assess the dynamics of enhancer activity during the early stages of murine ESC differentiation, we analyzed accessible genomic regions using STARR-seq, a massively parallel reporter assay. This resulted in a genome-wide quantitative map of active mESC enhancers, in pluripotency and during the early stages of differentiation. We find that only a minority of accessible regions is active and that such regions are enriched near promoters, characterized by specific chromatin marks, enriched for distinct sequence motifs, and modeling shows that active regions can be predicted from sequence alone. Regions that change their activity upon retinoic acid-induced differentiation are more prevalent at distal intergenic regions when compared to constitutively active enhancers. Further, analysis of differentially active enhancers verified the contribution of individual TF motifs toward activity and inducibility as well as their role in regulating endogenous genes. Notably, the activity of retinoic acid receptor alpha (RARα) occupied regions can either increase or decrease upon the addition of its ligand, retinoic acid, with the direction of the change correlating with spacing and orientation of the RARα consensus motif and the co-occurrence of additional sequence motifs. Together, our genome-wide enhancer activity map elucidates features associated with enhancer activity levels, identifies regulatory regions disregarded by computational prediction tools, and provides a resource for future studies into regulatory elements in mESCs.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
STARR-seq identifies active, chromatin-masked, and dormant enhancers in pluripotent mouse embryonic stem cells.Genome Biol. 2020 Sep 10;21(1):243. doi: 10.1186/s13059-020-02156-3. Genome Biol. 2020. PMID: 32912294 Free PMC article.
-
Comprehensive Genomic Discovery of Non-Coding Transcriptional Enhancers in the African Malaria Vector Anopheles coluzzii.Front Genet. 2022 Jan 10;12:785934. doi: 10.3389/fgene.2021.785934. eCollection 2021. Front Genet. 2022. PMID: 35082832 Free PMC article.
-
Functional Dissection of the Enhancer Repertoire in Human Embryonic Stem Cells.Cell Stem Cell. 2018 Aug 2;23(2):276-288.e8. doi: 10.1016/j.stem.2018.06.014. Epub 2018 Jul 19. Cell Stem Cell. 2018. PMID: 30033119 Free PMC article.
-
STARR-seq - principles and applications.Genomics. 2015 Sep;106(3):145-150. doi: 10.1016/j.ygeno.2015.06.001. Epub 2015 Jun 11. Genomics. 2015. PMID: 26072434 Review.
-
Principle and application of self-transcribing active regulatory region sequencing in enhancer discovery research.Yi Chuan. 2024 Aug;46(8):589-602. doi: 10.16288/j.yczz.24-149. Yi Chuan. 2024. PMID: 39140141 Review.
Cited by
-
A Mesenchymal stem cell Aging Framework, from Mechanisms to Strategies.Stem Cell Rev Rep. 2024 Aug;20(6):1420-1440. doi: 10.1007/s12015-024-10732-4. Epub 2024 May 10. Stem Cell Rev Rep. 2024. PMID: 38727878 Review.
-
RNA polymerase II dynamics shape enhancer-promoter interactions.Nat Genet. 2023 Aug;55(8):1370-1380. doi: 10.1038/s41588-023-01442-7. Epub 2023 Jul 10. Nat Genet. 2023. PMID: 37430091 Free PMC article.
-
An in vivo systemic massively parallel platform for deciphering animal tissue-specific regulatory function.Front Genet. 2025 Apr 9;16:1533900. doi: 10.3389/fgene.2025.1533900. eCollection 2025. Front Genet. 2025. PMID: 40270544 Free PMC article.
-
Emerging insights into enhancer biology and function.Transcription. 2023 Nov;14(1-2):68-87. doi: 10.1080/21541264.2023.2222032. Epub 2023 Jun 13. Transcription. 2023. PMID: 37312570 Free PMC article. Review.
-
Systematic mapping and modeling of 3D enhancer-promoter interactions in early mouse embryonic lineages reveal regulatory principles that determine the levels and cell-type specificity of gene expression.bioRxiv [Preprint]. 2023 Jul 19:2023.07.19.549714. doi: 10.1101/2023.07.19.549714. bioRxiv. 2023. Update in: Nat Struct Mol Biol. 2024 Jan;31(1):125-140. doi: 10.1038/s41594-023-01130-4. PMID: 37577543 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
