

Scaling-up a fragment-based protein-protein interaction method using a human reference interaction set
- PMID: 34850971
- PMCID: PMC9299658
- DOI: 10.1002/prot.26288
Scaling-up a fragment-based protein-protein interaction method using a human reference interaction set
Abstract
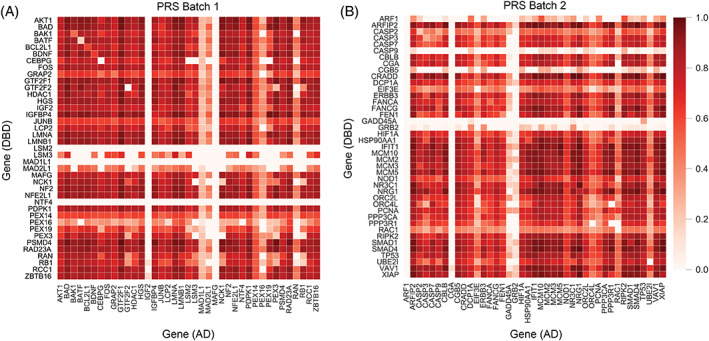
Protein-protein interactions (PPIs) are essential in understanding numerous aspects of protein function. Here, we significantly scaled and modified analyses of the recently developed all-vs-all sequencing (AVA-Seq) approach using a gold-standard human protein interaction set (hsPRS-v2) containing 98 proteins. Binary interaction analyses recovered 20 of 47 (43%) binary PPIs from this positive reference set (PRS), comparing favorably with other methods. However, the increase of 20× in the interaction search space for AVA-Seq analysis in this manuscript resulted in numerous changes to the method required for future use in genome-wide interaction studies. We show that standard sequencing analysis methods must be modified to consider the possible recovery of thousands of positives among millions of tested interactions in a single sequencing run. The PRS data were used to optimize data scaling, auto-activator removal, rank interaction features (such as orientation and unique fragment pairs), and statistical cutoffs. Using these modifications to the method, AVA-Seq recovered >500 known and novel PPIs, including interactions between wild-type fragments of tumor protein p53 and minichromosome maintenance complex proteins 2 and 5 (MCM2 and MCM5) that could be of interest in human disease.
© 2021 The Authors. Proteins: Structure, Function, and Bioinformatics published by Wiley Periodicals LLC.
Conflict of interest statement
The authors declare there are no competing interests.
Figures




Similar articles
-
Novel protein contact points among TP53 and minichromosome maintenance complex proteins 2, 3, and 5.Cancer Med. 2022 Dec;11(24):4989-5000. doi: 10.1002/cam4.4805. Epub 2022 May 14. Cancer Med. 2022. PMID: 35567389 Free PMC article.
-
Inferring high-confidence human protein-protein interactions.BMC Bioinformatics. 2012 May 4;13:79. doi: 10.1186/1471-2105-13-79. BMC Bioinformatics. 2012. PMID: 22558947 Free PMC article.
-
High-resolution protein-protein interaction mapping using all-versus-all sequencing (AVA-Seq).J Biol Chem. 2019 Jul 26;294(30):11549-11558. doi: 10.1074/jbc.RA119.008792. Epub 2019 Jun 10. J Biol Chem. 2019. PMID: 31182485 Free PMC article.
-
Interactome evolution: insights from genome-wide analyses of protein-protein interactions.Curr Opin Struct Biol. 2018 Jun;50:42-48. doi: 10.1016/j.sbi.2017.10.012. Epub 2017 Nov 5. Curr Opin Struct Biol. 2018. PMID: 29112911 Review.
-
Utility of next-generation RNA-sequencing in identifying chimeric transcription involving human endogenous retroviruses.APMIS. 2016 Jan-Feb;124(1-2):127-39. doi: 10.1111/apm.12477. APMIS. 2016. PMID: 26818267 Review.
Cited by
-
Novel protein contact points among TP53 and minichromosome maintenance complex proteins 2, 3, and 5.Cancer Med. 2022 Dec;11(24):4989-5000. doi: 10.1002/cam4.4805. Epub 2022 May 14. Cancer Med. 2022. PMID: 35567389 Free PMC article.
-
Identifying novel interactions of the colon-cancer related APC protein with Wnt-pathway nuclear transcription factors.Cancer Cell Int. 2022 Dec 1;22(1):376. doi: 10.1186/s12935-022-02799-1. Cancer Cell Int. 2022. PMID: 36457029 Free PMC article.
-
Long-read sequencing to detect full-length protein-protein interactions.Sci Rep. 2025 Jul 17;15(1):25967. doi: 10.1038/s41598-025-08549-3. Sci Rep. 2025. PMID: 40676061 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
