

Autocrine inhibition by a glutamate-gated chloride channel mediates presynaptic homeostatic depression
- PMID: 34851664
- PMCID: PMC8635443
- DOI: 10.1126/sciadv.abj1215
Autocrine inhibition by a glutamate-gated chloride channel mediates presynaptic homeostatic depression
Abstract
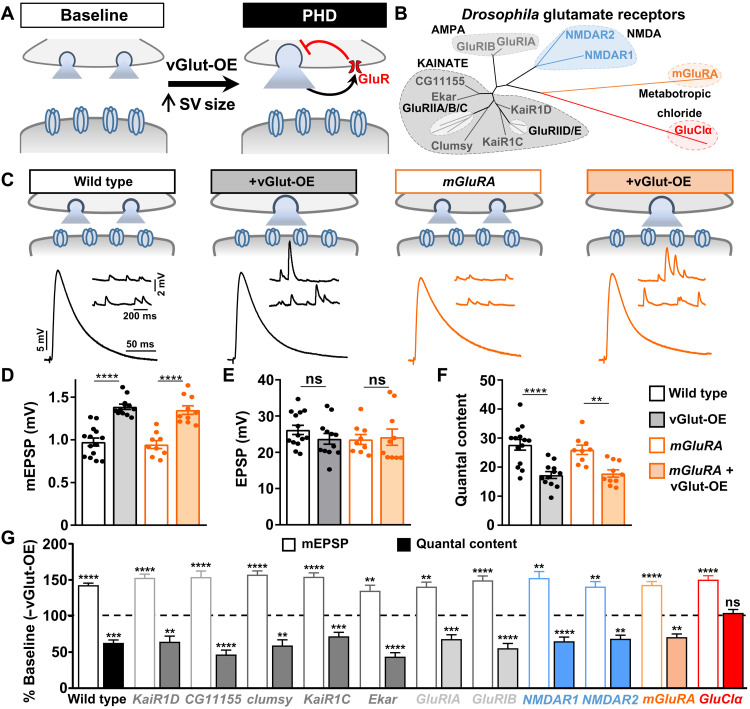
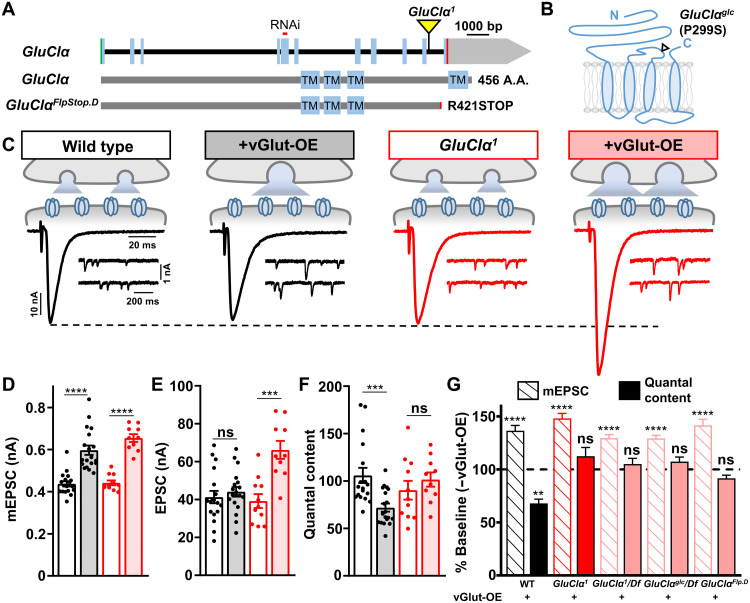
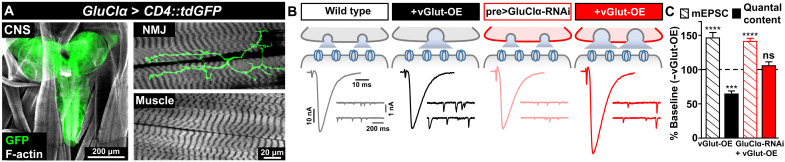
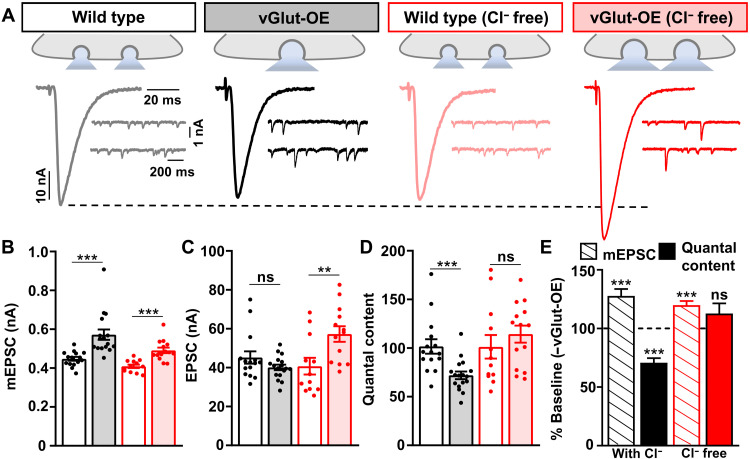
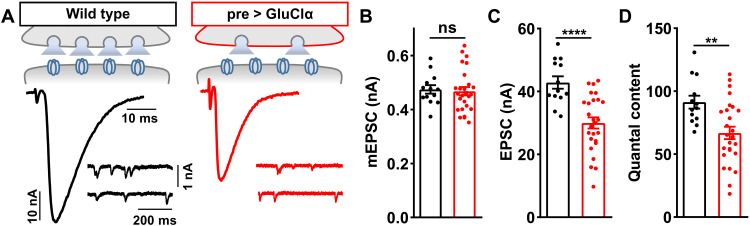
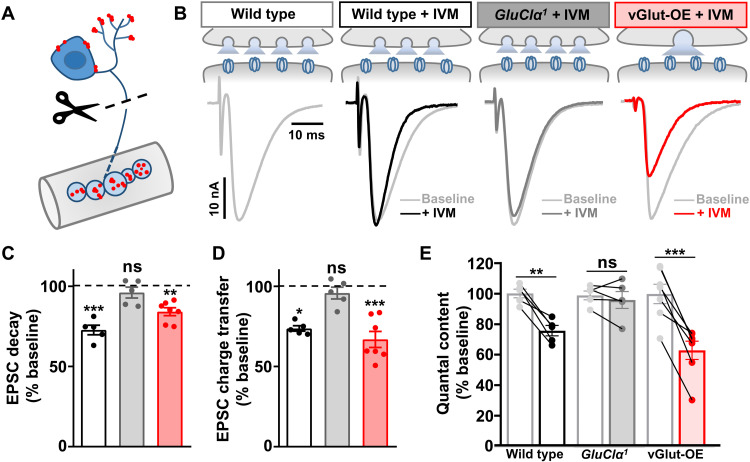
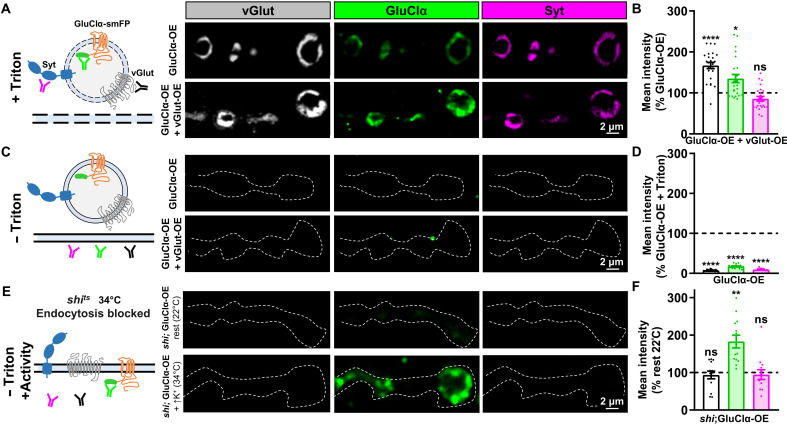
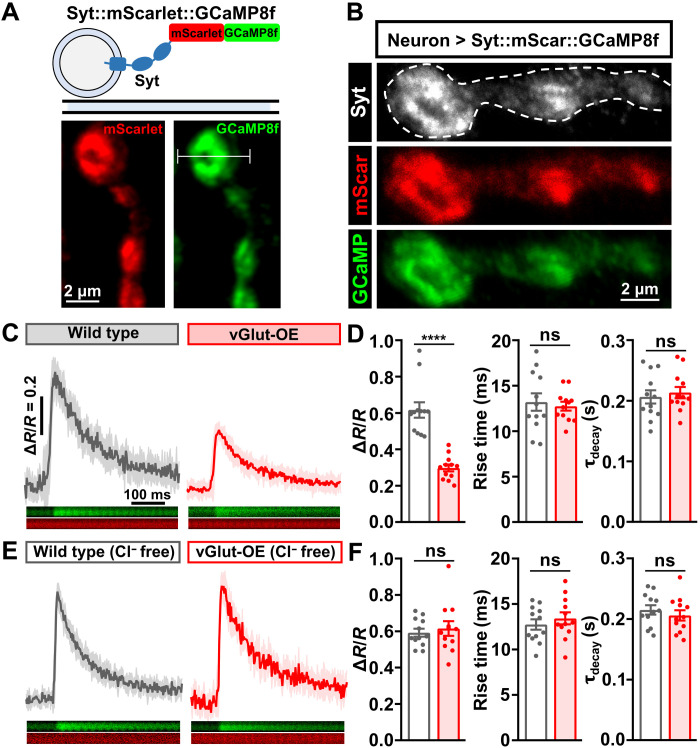
Homeostatic modulation of presynaptic neurotransmitter release is a fundamental form of plasticity that stabilizes neural activity, where presynaptic homeostatic depression (PHD) can adaptively diminish synaptic strength. PHD has been proposed to operate through an autocrine mechanism to homeostatically depress release probability in response to excess glutamate release at the Drosophila neuromuscular junction. This model implies the existence of a presynaptic glutamate autoreceptor. We systematically screened all neuronal glutamate receptors in the fly genome and identified the glutamate-gated chloride channel (GluClα) to be required for the expression of PHD. Pharmacological, genetic, and Ca2+ imaging experiments demonstrate that GluClα acts locally at axonal terminals to drive PHD. Unexpectedly, GluClα localizes and traffics with synaptic vesicles to drive presynaptic inhibition through an activity-dependent anionic conductance. Thus, GluClα operates as both a sensor and effector of PHD to adaptively depress neurotransmitter release through an elegant autocrine inhibitory signaling mechanism at presynaptic terminals.
Figures
Similar articles
-
A Glutamate Homeostat Controls the Presynaptic Inhibition of Neurotransmitter Release.Cell Rep. 2018 May 8;23(6):1716-1727. doi: 10.1016/j.celrep.2018.03.130. Cell Rep. 2018. PMID: 29742428 Free PMC article.
-
Endogenous Tagging Reveals Differential Regulation of Ca2+ Channels at Single Active Zones during Presynaptic Homeostatic Potentiation and Depression.J Neurosci. 2019 Mar 27;39(13):2416-2429. doi: 10.1523/JNEUROSCI.3068-18.2019. Epub 2019 Jan 28. J Neurosci. 2019. PMID: 30692227 Free PMC article.
-
The auxiliary glutamate receptor subunit dSol-1 promotes presynaptic neurotransmitter release and homeostatic potentiation.Proc Natl Acad Sci U S A. 2020 Oct 13;117(41):25830-25839. doi: 10.1073/pnas.1915464117. Epub 2020 Sep 24. Proc Natl Acad Sci U S A. 2020. PMID: 32973097 Free PMC article.
-
Homeostatic control of presynaptic neurotransmitter release.Annu Rev Physiol. 2015;77:251-70. doi: 10.1146/annurev-physiol-021014-071740. Epub 2014 Nov 5. Annu Rev Physiol. 2015. PMID: 25386989 Review.
-
Homeostatic plasticity at the Drosophila neuromuscular junction.Neuropharmacology. 2014 Mar;78:63-74. doi: 10.1016/j.neuropharm.2013.06.015. Epub 2013 Jun 24. Neuropharmacology. 2014. PMID: 23806804 Free PMC article. Review.
Cited by
-
Distinct input-specific mechanisms enable presynaptic homeostatic plasticity.bioRxiv [Preprint]. 2024 Sep 12:2024.09.10.612361. doi: 10.1101/2024.09.10.612361. bioRxiv. 2024. Update in: Sci Adv. 2025 Feb 14;11(7):eadr0262. doi: 10.1126/sciadv.adr0262. PMID: 39314403 Free PMC article. Updated. Preprint.
-
Ca2+ imaging of synaptic compartments using subcellularly targeted GCaMP8f in Drosophila.STAR Protoc. 2024 Mar 15;5(1):102832. doi: 10.1016/j.xpro.2023.102832. Epub 2024 Jan 9. STAR Protoc. 2024. PMID: 38198278 Free PMC article.
-
Botulinum neurotoxin accurately separates tonic vs. phasic transmission and reveals heterosynaptic plasticity rules in Drosophila.Elife. 2022 Aug 22;11:e77924. doi: 10.7554/eLife.77924. Elife. 2022. PMID: 35993544 Free PMC article.
-
Circadian Vesicle Capture and Phase-Delayed Activity Maximize Synaptic Neuropeptide Release by Clock Neurons.bioRxiv [Preprint]. 2025 Feb 25:2023.12.01.569590. doi: 10.1101/2023.12.01.569590. bioRxiv. 2025. PMID: 38106047 Free PMC article. Preprint.
-
Molecular organization of central cholinergic synapses.Proc Natl Acad Sci U S A. 2025 Apr 29;122(17):e2422173122. doi: 10.1073/pnas.2422173122. Epub 2025 Apr 24. Proc Natl Acad Sci U S A. 2025. PMID: 40273107 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
