

Emergence and global spread of Listeria monocytogenes main clinical clonal complex
- PMID: 34851675
- PMCID: PMC8635441
- DOI: 10.1126/sciadv.abj9805
Emergence and global spread of Listeria monocytogenes main clinical clonal complex
Abstract
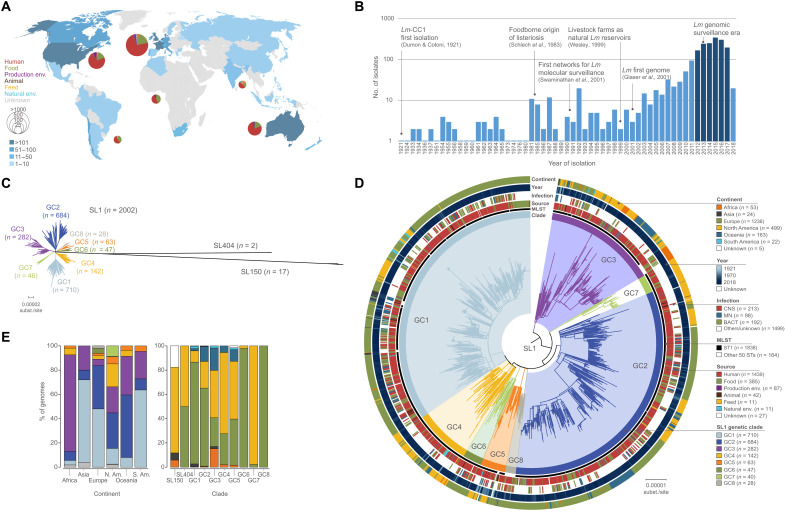
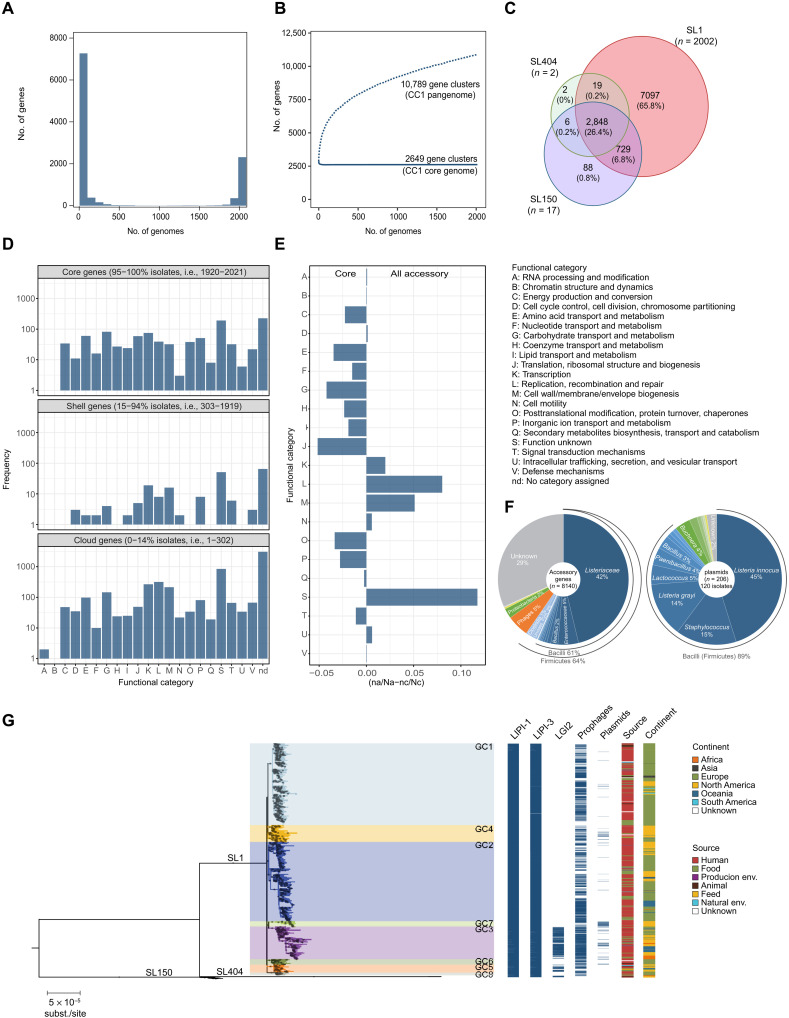
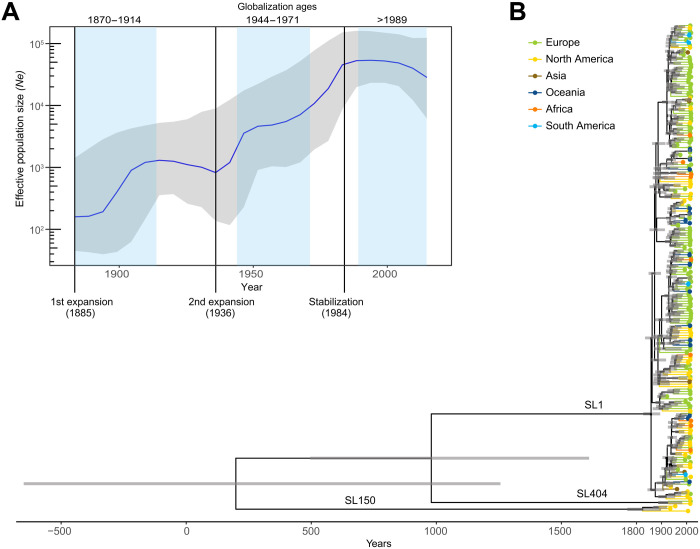
The bacterial foodborne pathogen Listeria monocytogenes clonal complex 1 (Lm-CC1) is the most prevalent clonal group associated with human listeriosis and is strongly associated with cattle and dairy products. Here, we analyze 2021 isolates collected from 40 countries, covering Lm-CC1 first isolation to present days, to define its evolutionary history and population dynamics. We show that Lm-CC1 spread worldwide from North America following the Industrial Revolution through two waves of expansion, coinciding with the transatlantic livestock trade in the second half of the 19th century and the rapid growth of cattle farming and food industrialization in the 20th century. In sharp contrast to its global spread over the past century, transmission chains are now mostly local, with limited inter- and intra-country spread. This study provides an unprecedented insight into L. monocytogenes phylogeography and population dynamics and highlights the importance of genome analyses for a better control of pathogen transmission.
Figures


References
-
- Swaminathan B., Gerner-Smidt P., The epidemiology of human listeriosis. Microbes Infect. 9, 1236–1243 (2007). - PubMed
-
- Charlier C., Perrodeau É., Leclercq A., Cazenave B., Pilmis B., Henry B., Lopes A., Maury M. M., Moura A., Goffinet F., Dieye H. B., Thouvenot P., Ungeheuer M.-N. N., Tourdjman M., Goulet V., de Valk H., Lortholary O., Ravaud P., Lecuit M.; MONALISA Study Group , Clinical features and prognostic factors of listeriosis: The MONALISA national prospective cohort study. Lancet Infect. Dis. 17, 510–519 (2017). - PubMed
-
- Orsi R. H., den Bakker H. C., Wiedmann M., Listeria monocytogenes lineages: Genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 301, 79–96 (2011). - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
